All published articles of this journal are available on ScienceDirect.

In Silico Study of Pubchem Compounds for Solanum torvum as Antiviral Agent against SARS-CoV-2
Authors Info & Affiliations
Abstract
Background:
The recent epidemic outbreak of a novel coronavirus called SARS-CoV-2 has caused suffering among many people in the form of respiratory tract infection. Currently, there are no targeted drugs, and effective treatment options remain limited.
Objective:
In order to rapidly discover new compounds for clinical purposes, in silico drug design and virtual drug screening have been initiated to identify new drug leads that target the main protease of the COVID-19 virus. Mpro is a key CoV enzyme, which plays a pivotal role in mediating viral replication and transcription, making it an attractive drug target for this virus.
Methods:
The present study was done to investigate the PubChem compounds of an ayurvedic herb Solanum torvum as an effective antiviral agent against COVID-19. The PubChem compounds like Torvoside H, Torvoside A, Torvoside E, Torvoside F, Torvonin A, 2,3,4-trimethyltriacontane, Torvanol A Q27134802, 5-hexatriacontanone, Jurubine, Tritriacontan-3-one, Torvanol A, Chlorogenone Spirostane-3,6-dione of Solanum torvum were downloaded from NCBI PubChem database acting as ligands for protein ligand docking. The 3D structure of the viral MPro (PDB ID: 6yb7) was retrieved from the RCSB PDB database. The active sites and binding sites were analyzed, and Docking molecular simulations were realized among a total of 12 ligands against COVID-19.
Results:
The PubChem compounds from the fruits of Solanum torvum showed good docking score and protein-ligand interaction, indicating that the PubChem compounds can cure the COVID-19 disease and act as an effective antiviral agent.
Conclusion:
Most of the PubChem compounds in the fruits of Solanum torvum showed better paramagnetic parameters.
1. INTRODUCTION
Towards the end of the year 2019, an outbreak of pneumonia of unknown cause emerged from the Hubei province of China, the city of Wuhan being the epicenter [1]. In a short time, Chinese authorities rapidly isolated and characterized a novel coronavirus closely related to the SARS-CoV that caused the outbreak of a severe acute respiratory syndrome 18 years ago in China [2]. The virus named severe acute respiratory syndrome–coronavirus-2 (SARS-CoV-2) has an RNA genome that shares about 82% similarity with that of the SARS coronavirus (SARS-CoV). Both the viruses belong to the clade b of the genus Betacoronavirus [3, 4]. The disease caused by SARS-CoV-2 is called coronavirus disease 2019 (COVID-19). Initially, at the beginning of the outbreak, the emergence was related to the Hunan seafood and animal market in Wuhan; efficient human-to-human transmission led to exponential growth in the number of cases. One of the best-characterized drug targets among coronaviruses is the main protease (Mpro, also called 3CLpro [5]. Along with the papain-like protease(s), this enzyme is essential for processing the polyproteins that are translated from the viral RNA [6]. The Mpro operates at no fewer than 11 cleavage sites on the large polyprotein 1ab (replicase 1ab, ~790 kDa); the recognition sequence at most sites is Leu-Gln↓(Ser, Ala, Gly). With no clear method of cure through chemotherapy in sight, the only dependable mode is through traditional medicines.
Traditional medicine is ‘the knowledge, skills, and practices based on the theories, beliefs, and experiences indigenous to different cultures, used in the maintenance of health and the prevention, diagnosis, improvement or treatment of physical and mental illness” [7]. The main aim of traditional medicine is to use the resources available from nature for the treatment of diseases and to maintain the goodness of life. Solanum torvum Sw. (Family: Solanaceae), commonly known as Turkey Berry is native to Mexico, Peru, and Venezuela [8]. It is widely distributed in Africa, West Indies, India, Bermuda, Indonesia, Malaya, China, the Philippines, and tropical America [9]. Studies done on Solanum torvum have shown that the plant possesses significant antimicrobial, anticancer, diuretic, anti-inflammatory, anti-influenza activity [10]. It is used in folk and traditional medicine for the treatment of diseases like asthma, diabetes, gastrointestinal diseases, etc. In fact, around 2000 years ago a Tamil saint Agasthiyar reported Solanum torvumto be a cure for respiratory tract infection through the medicinal palm script of the plant [11]. Torvanol A, torvoside H, and compounds aglycone torvoside H isolated from Solanum torvum exhibited antiviral activity (herpes simplex virus type1) with the IC50 values of 9.6, 23.2, and 17.4µg/ml, respectively [12]. Asthma is a common long-term inflammatory disease of the airways of the lungs. It is characterized by recurring symptoms like wheezing, coughing, chest tightness, and shortness of breath. These symptoms are due to reversible airflow obstruction and bronchospasm [13]. Treatment with glucocorticoids has been consistently shown to reduce not only the symptoms of asthma but also bronchial hyperresponsiveness [14]. In contrast to the rapid inhibitory effects of β2- agonists, glucocorticoids given in a single dose are not effective in preventing early allergen-invoked bronchoconstriction, but inhibition of the late response has been clearly demonstrated [15]. The inhaled glucocorticoids consistently reduce hyper airway reactivity in asthmatics [16]. From the docking and drug-likeness study, compounds such as Cholesta-5,7,9-(11)-trien-3-ol,4,4-dimethyl, (3a); Lanosta-7,9-(11),20-triene-3α, 18-diol, diacetate and Cholestan-26-oic acid,3,7,12,24-tetrakis (acetyloxy), methyl ester, (3a,5a,7a,12a) were found to have the affinity of-6.8,-6.9 and-6.9 towards Gr which is very similar to the affinity of the standard (- 7.1) [17-19].
The development of a potential drug for COVID-19 will need over 15 years from target identification, target validation, hit discovery, lead optimization, and preclinical and clinical trials [20]. Therefore, it is virtually not possible to develop de novo drugs in the time frame needed to impact the current SARS-CoV-2 crisis. Hence, the most feasible approach is to find potential cures from clinical drugs by drug repurposing (also known as drug repositioning, reprofiling, redirecting, or rediscovering). Drug repurposing can rapidly expand target disease indications of an existing drug while saving time and money since the data for human pharmacokinetics, safety, and preclinical results are already available [21]. Successful examples of drug repurposing include the use of sildenafil in treating erectile dysfunction and the anti-cancer uses of thalidomide [22]. The fastest way for the development of medicines treating SARS-CoV- 2 is to find potential molecules from the marketed drugs [23]. Another research discovered that carfilzomib, eravacycline, valrubicin, elbasvir, streptomycin are very promising to have an affinity of interaction with the receptor SARS-CoV-2 [24].
Recent research concluded that paritaprevir and simeprevir were identified as potential inhibitors of SARS-CoV-2 3CLpro [25]. At the Rajavithi Hospital in Thailand, the infectious disease team used a combination of oseltamivir (anti-influenza agent) and lopinavir plus ritonavir (HIV protease inhibitors) to successfully improve the condition of the patients with severe conditions [26]. The antiviral effects of remdesivir have been assessed in humans, and it has been used to treat severe COVID-19 patients as an emergency use [27-29]. However, further studies are required to assess the effectiveness and safety of remdesivir for COVID-19 [30].
2. MATERIALS AND METHODS
2.1. Preparation of Receptors
The crystallographic structure of the main protease SARS-CoV- 2 (PDB ID: 6lu7) used in this work presents a high resolution of 2:16Å, contains a total of 306 residues, and has a molecular mass of approximately 31:4kDa Fig. (S1). This crystal structure, originally resolved by Liu et al., was retrieved from the Protein Data Bank (PDB) deposited in January 2020 [31].
2.2. Prediction of Binding Sites or Active Sites
The active site, binding sites, and amino acid residues in 6lu7 was identified using an online program metaPocket 2.0 (http://projects.biotec.tu-dresden.de/metapocket). MetaPocket 2.0 is a meta server to identify ligand binding sites on protein surface it is a consensus method, in which the predicted binding sites from eight methods like LIGSITECS, PASS. QSiteFinder, SURFNET, Fpocket, GHECOM, ConCavity, and POCASA are combined together to predict the sites.
2.3. Preparation of Ligands
The 2D structures of the phytochemical compounds of fruits of Solanum torvum were downloaded from PubChem (https://pubchem.ncbi.nlm.nih.gov) in SDF format and then converted into 3D PDB format using Open Babel software.
2.4. Drug-Likeness Activity Prediction
All the Pubchem compounds were tested for the drug-likeness through the DruLiTo software, which is open-source software. The pharmacological significance of a ligand is evaluated on various properties like drug bioavailability, drug-likeness or ADMET, etc. These properties are calculated using certain Physico-chemical and structural properties. Therefore, all ligands were evaluated for their drug-like nature under Lipinski’s rules of five8 by DruLiTo software.
2.5. In Silico Molecular Docking
Main protease of COVID-19, Mpro, PDB ID: 6lu7 receptor was docked with different ligands using Hex 8.0.0 docking software. Hex docking was carried out by setting appropriate parameters such as twist range-360, receptor range-180, FFT mode-3D fast lite, ligand range-180, grid dimension-0.6, and distance range-40. The binding energy produced by docking action was tabulated.
2.6. Protein-ligand Interaction Analysis
The Protein-Ligand Interaction Profiler (PLIP), a novel web service for fully automated detection and visualization of relevant non-covalent protein-ligand contacts in 3D structures (projects.biotec.tu-dresden.de/plip-web). It detects interactions on a single atom level, covering seven interaction types (hydrogen bonds, hydrophobic contacts, pi-stacking, pi-cation interactions, salt bridges, water bridges, and halogen bonds). All the docked protein-ligand files were analyzed by using the PLIP tool.
2.7. Structural Analysis and Visualization
For structural analysis and visualization, UCSF Chimera was used. UCSF Chimera, often called Chimera, is a noncommercial free web resource developed by the University of California, San Francisco, to make changes and allow the addition of new abilities for effective functioning. It is a facility for interactive visualization and analysis of molecular structures. It can also be used to analyze the data related to the results obtained by docking and their trajectories.
3. RESULTS
3.1. Virtual Screening
Bioinformatics is emerging as an important tool in the field of pharmaceutical and drug development with the prospective to significantly improvise the drugs and check how they are originated, carried to the clinical trials, and eventually released to the marketplace. This study was performed using covid19 protein and twelve different compounds; the receptor was derived from Protein Data Bank (PDB ID: 6lu7) and was used as a target for stimulating docking against the twelve different compounds. The PDB file of the protein was opened in UCSF Chimera version 1.12, and all non-standard atoms and bonds were removed along with the receptor involved for protein-ligand docking using hex software (Fig. S1).
The active site of the 6lu7 protein was analyzed by MetaPocket 2.0 server. The Six pocket sites and six ligand binding sites were significantly recorded, and the sites acted as binding areas for Solanum torvum ligands (Fig. S2). Solanum torvum ligands were retrieved from NCBI Pubchem database. The twelve Pubchem compounds are Torvoside H (25S)-26-O-(beta-D-glucopyranosyl)-6alpha,26-dihydroxy-5alpha-spiros tan-3-one 6-O-[alpha-L-rhamnopyranosyl-(1->3)-beta-D-quin ovopyranoside (C45H74O18), 5-hexatriacontanone (C36H72O), Jurubine (C33H57NO8), Torvoside E (C40H66O14), Torvoside F (C45H74O18), Torvonin A(C39H64O12), 2,3,4-trimethyltriacontane (C33H68), Torvoside A (C45H76O18), Tritriacontan-3-one (C33H66O), Torvanol A (C20H20O10S), Chlorogenone, Spiros tane-3,6-dione (C27H40O4), Torvanol A Q27134802 rel-potassium (3R,4S)-3-{5-[(E)-2-carboxyethenyl]-2-hydroxy-3-methoxyphenyl}-6-methoxy-3,4-dihydro-2H-chromen-4-yl sulfate (C20H19KO10S). The PubChem ID, compound name, molecular formula, and molecular weight are shown in Table S1.
3.2. Drug-Likeness Activity Prediction
The results of DruLiTo software shows that the molecular weight (Mw) of Pubchem compounds are under 500, partition coefficient (log P) value is under 5, octanol-water partition coefficient (AlogP), the numbers of hydrogen bond acceptors (HBA) are under 10, and several hydrogen bonds are donors (HBD) under 5.27 phytochemicals, total polar surface area (TPSA), atom molar refractivity (AMR), number of rotatable bonds (nRB), rotatable bond count (RC), number of rigid bonds (nRigidB), number of aromatic ring and number of hydrogen bonds (nHB), were found to agree with Lipinski’s rule of 5. Hence, the Pubchem compounds follow Lipinski’s rule of 5. However, four Pubchem compounds of Solanum torvum showed significantly excellent results like Torvoside H (25S)-26-O-(beta-D-glucopyranosyl)-6alpha,26-dihydroxy-5 alpha-spirostan-3-one 6-O-[alpha-L-rhamnopyranosyl-(1->3)-beta-D-quinovopyranoside (C45H74O18) showed the value of logP (1.229), AlogP (-5.084), HBA (18), HBD (10), TPSA (283.98), AMR (212.14), number of rigid bonds (59) and number of hydrogen bonding (28). Also, Torvanol A (C20H20O10S) recorded value of log P (0.513), A log P (-0.674), HBA (10), HBD (3), TPSA (52.6), AMR (114.74), number of rigid bonds (26), number of the aromatic ring (2) and number of hydrogen bonding (13) respectively. Torvanol A Q27134802 rel-potassium (3R,4S)-3-{5-[(E)-2-carboxyethenyl]-2-hydroxy- 3-methoxyphenyl}-6-methoxy-3,4-dihydro-2H-chromen-4-yl sulfate (C20H19KO10S) also showed significant result and torvoside F (C45H74O18) showed the value of log P (1.288), AlogP (-4.665), HBA(18), HBD(9), TPSA(272.98), AMR(212.9), number of rigid bond(58) and number of hydrogen bonding (27). Similarly Trovoside A (C45H76O18) recorded value of logP (1.638), AlogP (-5.433), HBA (18), HBD (11), TPSA (287.14), AMR (211.02), number of rigid bonds (59), and number of hydrogen bonding (29) (Table 1). Jurubine (C33H57NO8), Torvoside E (C40H66O14), Torvonin A (C39H64O12), and Chlorogenone Spirostane-3,6-dione (C27H40O4) showed moderate results like logP value range between 3.011 to 5.113, AlogP (-2.46 to 0.111) HBA (4 -14), HBD (0 - 7) TPSA(56.6 - 214), AMR (114.4 – 185.8) number of rigid bond (36 - 54) and a number of hydrogen bonding (4 – 21) significantly gives adequate bioavailability. Compounds such as 5-hexatriacontanone (C36H72O), 2,3,4-trimethyltriacontane (C33H68), and Tritriacontan-3-one (C33H66O) showed inferior results as these compounds contain significant medical properties. This proved our finding that the plant Solanum torvum contains several phytochemical compounds. Through this study, it has been determined that the maximum number of the compound had significant drug ability while some results showed minimum drug activity.
3.3. Molecular Docking Studies
The intermolecular flexible docking simulation was performed to explore the binding site of the twelve PubChem compounds with SARS-CoV- 2 (PDB ID: 6lu7). Energy values were calculated from the information obtained while docking the conformations of the receptor-ligand complex. The crucial interaction information concerning the binding sites and their orientation of the target protein was obtained. The binding affinity was measured in terms of KJ/mol. The results of docking and crucial interaction between the ligand and the receptor are shown in Table 2.
| S.No. | Compound | Mw g/mol | logP | AlogP | HBA | HBD | TPSA | AMR | nRB | No. Atom | RC | No. Rigid Bond | No. Arom Ring | nHB | |||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1. | Torvoside H | 903.1 | 1.229 | -5.084 | 18 | 10 | 283.98 | 212.14 | 11 | 137 | 8 | 59 | 1 | 28 | |||
| 2. | Torvonin A | 724.9 | 4.193 | -2.919 | 12 | 6 | 176.76 | 175.1 | 4 | 115 | 8 | 54 | 0 | 18 | |||
| 3. | Torvoside E | 770.9 | 3.011 | -3.428 | 14 | 7 | 214.06 | 185.8 | 10 | 120 | 7 | 50 | 0 | 21 | |||
| 4. | Torvoside A | 905.1 | 1.638 | -5.433 | 18 | 11 | 287.14 | 211.02 | 11 | 139 | 8 | 59 | 0 | 29 | |||
| 5. | 2,3,4-trimethyltriacontane | 464.9 | 18.93 | -5.765 | 0 | 0 | 0 | 111.5 | 27 | 101 | 0 | 5 | 0 | 0 | |||
| 6. | Torvanol A | 490.5 | -0.513 | nan | 10 | 2 | 160.03 | nan | 7 | 51 | 3 | 26 | 2 | 12 | |||
| 7. | 5-hexatriacontanone | 521 | 10.33 | -9.02 | 1 | 0 | 17.07 | 116.5 | 33 | 109 | 0 | 5 | 0 | 1 | |||
| 8. | Torvanol A | 452.4 | -0.513 | -0.674 | 10 | 3 | 157.2 | 114.74 | 7 | 51 | 3 | 26 | 2 | 13 | |||
| 9. | Jurubine | 595.8 | 5.113 | -2.46 | 9 | 6 | 154.8 | 149.1 | 7 | 99 | 6 | 40 | 0 | 14 | |||
| 10. | Tritriacontan-3-one | 478.9 | 16.623 | -8.153 | 1 | 0 | 17.07 | 107.77 | 30 | 100 | 0 | 3 | 0 | 1 | |||
| 11. | Torvoside F | 903.1 | 1.288 | -4.665 | 18 | 9 | 272.98 | 212.9 | 12 | 137 | 8 | 58 | 0 | 27 | |||
| 12. | Chlorogenone, Spirostane-3,6-dione |
428.6 | 4.61 | 0.111 | 4 | 0 | 52.6 | 114.42 | 0 | 71 | 6 | 36 | 0 | 4 | |||
| S. NO. | Ligand | COVID -19 (Mpro) PDB ID: | Eforce (binding energy of ligand) | Docking score ETotal Value KJ/mol | Eshape (energy content of the protein) |
|---|---|---|---|---|---|
| 1. | Torvoside H | 6lu7 | 0 | -399.7 | -399.7 |
| 2. | Torvonin A | 6lu7 | 0 | -394.5 | -394.5 |
| 3. | Torvoside E | 6lu7 | 0 | -386.4 | -386.4 |
| 4. | Torvoside A | 6lu7 | 0 | -351 | -351 |
| 5. | 2,3,4-trimethyltriacontane | 6lu7 | 0 | -339.3 | -339.3 |
| 6. | Torvanol A | 6lu7 | 0 | -330.0 | -330.0 |
| 7. | 5-hexatriacontanone | 6lu7 | 0 | -329.5 | -329.5 |
| 8. | Torvanol A | 6lu7 | 0 | -309.8 | -309.8 |
| 9. | Jurubine | 6lu7 | 0 | -286.9 | -286.9 |
| 10. | Tritriacontan-3-one | 6lu7 | 0 | -285.1 | -285.1 |
| 11. | Torvoside F | 6lu7 | 0 | -260..7 | -260..7 |
| 12. | Chlorogenone, Spirostane-3,6-dione |
6lu7 | 0 | -253 | -253 |
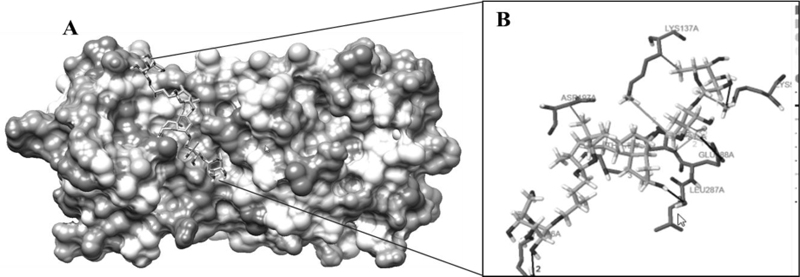
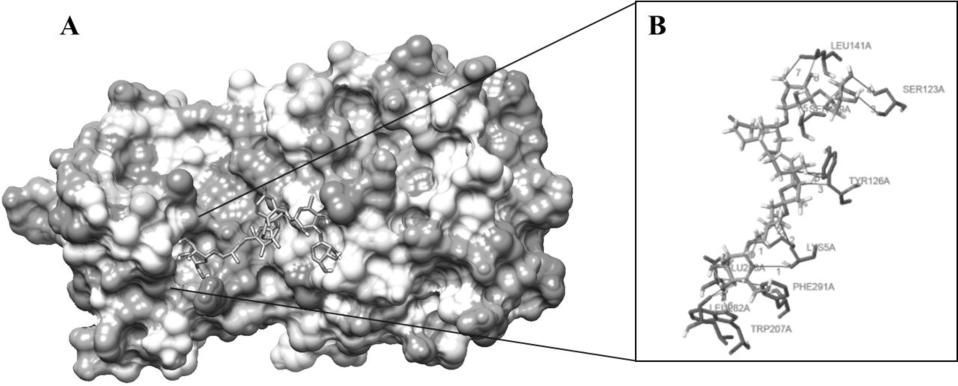
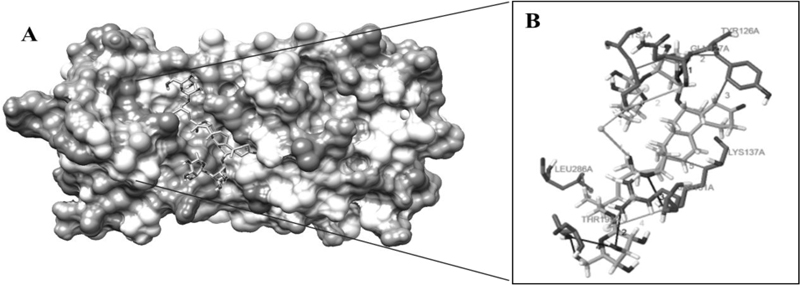
The highest negative energy value obtained after docking among twelve compounds was found between Torvoside H(25S)-26-O-(beta-D-glucopyranosyl)-6alpha,26-dihydroxy-5 alpha-spirostan-3-one6-O-[alpha-L-rhamnopyranosyl-(1->3)-beta-D-quinovopyranoside (C45H74O18) and 6lu7 (Fig. 1) value of -399.7, Torvonin A with 6lu7 (Fig. 2) with value -394.5, followed by Torvoside E with 6lu7 (Fig. 3), Torvoside A with 6lu7 (Fig. S3), 2,3,4-trimethyltriacontane with 6lu7 (Fig. S4), Torvanol A Q27134802 rel-potassium (3R,4S)-3-{5-[(E)-2-carboxyethenyl]-2-hydroxy-3-methoxyphenyl}-6-meth oxy-3,4-dihydro-2H-chromen-4-yl sulfate with 6lu7 (Fig. S5), 5 hexatriacontanone with 6lu7 (Fig. S6), Torvanol A with 6lu7 (Fig. S7), Jurubine with 6lu7 (Fig. S8), Tritriacontan-3-one with 6lu7 (Fig. S9), and Torvoside F with 6lu7 (Fig. S10), with docking scores -386.4, -351, -339.3, -330.0, -329.5, -309.8, -286.9, -285.1 and -260.7, respectively. While the least docking score was in docking of Chlorogenone, Spirostane-3,6-dione with 6lu7 (Fig. S11) and the scoring was -253 using Hex 8.0.0 docking software. This docking result analysis showed most of the ligand compounds had a high negative score. The higher the negative E total value, the stronger the interaction between ligand and receptor is, leading to activation of receptors. This docking result showed that each ligand binds with unique active sites of receptor protein and all the Pubchem compounds docked with COVID-19 receptor protein and all the ligands have unique medicinal properties.
3.4. Molecular Docking and Protein-Ligand Interactions Analysis
Protein-ligand interactions, including hydrogen bonding interactions, hydrophobic interaction, Water Bridge, salt bridges, and π-π interactions, showed significant results. Torvoside H (25S)-26-O-(beta-D-glucopyranosyl)-6alpha,26-dihydroxy-5alpha-spirostan-3-one 6-O-[alpha-L-rhamnopyrano syl-(1->3)-beta-D-quinovopyranoside (C45H74O18) recorded highest docking score with 6lu7. LYS5A, TYR126A, TYR126A, GLN127A, LYS137A, and LEU286A of the amino acid residues hydrophobic interacted with the ligand atom numbers 3072, 3072, 3058, 3072, 3047, and 3070 of Torvoside H. The bonding distances were calculated as 3.77, 3.34, 3.33, 3.34, 3.82, and 3.69. The hydrogen bonds between the amino acid position at GLU127A, ARG131A, LYS137A, THR199A, THR199A, and THR199A with ligand atom at 3026(O3), 3036(O3), 3022(O3), 1865(O3), 3039(O3), and 1865(O3) the bond distance of hydrogen acceptor at 3029, 2.63, 2.60, 3.00, 2.67 and 2.41 were recorded high interaction (Table S2 and Fig. 1). Torvonin A (C39H64O12) exhibited the best docking score with 6lu7. LYS137A, ASP197A, and THR199A of the amino acid residues hydrophobic interacted with the ligand atom numbers 3083, 3054, and 3056 of the Torvonin A. The bonding distances were calculated as 2.71, 3.06, and 3.98, respectively. The hydrogen bonds between the amino acid position at LYS5A, LYS236A, LUE287A, and GLU288A with ligand atom at 3033 (O3), 3034 (O3), 3025 (O3), and 2706 (O3) with the bonding distance of hydrogen acceptor at 3.28, 3.07, 2.57 and 3.49 were showed significant interaction. Water and salt bridges were analyzed in docked molecules. (Table S3 and Fig. 2). Torvoside E (C40H66O14) showed a significant docking score of 6lu7. LYS5A, LYS5A, TYR126A, TYR126A, TYR126A, LUA141A, LUA141A, GLU288A, and PHE291A of the amino acid residues interacting with the ligand atom numbers 3063, 3071, 3043, 3047, 3040, 3072, 3073, 3083, and 3083 of the Torvoside E. The bonding distances were calculated as 2.23, 3.25, 2.95, 3.20, 3.75, 3.54, 2.91, 3.92 and 3.93 respectively. The hydrogen bonds, water, and salt bridges were analyzed in docked molecules (Table S4 and Fig. 3). Torvoside A (C45H76O18) showed the best docking score with 6lu7. VAL104A, VAL104A, ASN151A, THR292A, and PHE294A of the amino acid residues interacted with the ligand atom numbers 3069, 3053, 3051, 3067, and 3055 of the Torvoside A. The bonding distances were calculated as 3.72, 3.75, 2.89, 3.31, and 3.37, respectively. The hydrogen bonds and water bridges were analyzed in docked molecules (Table S5 and Fig. S3). 2,3,4-trimethyltriacontane (C33H68) showed an elite docking score of 6lu7. ARG4A, LYS5A, and PHE291A of the amino acid residues interacting with the ligand atom numbers 3027, 3029and 3036 of the ligand with the bond distance of 3.63, 2.71, and 3.33 (Table S6 and Fig. S4).
Torvanol A Q27134802 rel-potassium (3R,4S)-3-{5-[(E)-2-carboxyethenyl]-2-hydroxy-3-methoxyphenyl}-6-metho xy-3,4-dihydro-2H-chromen-4-yl sulfate (C20H19KO10S), 5-hexatriacontanone (C36H72O) showed best docking score with 6lu7. ARG4A, LYS5A, LYS5A, LYS137A, and LUE286A of the amino acid residues interact with the ligand atom numbers 3052, 3044, 3043, 3028, and 3033 of the ligand with the bond distance of 3.66, 3.43, 3.48, 3.61and 3.78. (Table S7 and Fig. S5). 5-hexatriacontanone (C36H72O) showed the best docking score with 6lu7. LYS137A, ASP197A, and LUE286A of the amino acid residues interacted with the ligand atom numbers 3072, 3051, and 3049 of the ligand with the bond distance of 3.07, 2.86, and 3.76 (Table S8 and Fig. S6). Torvanol A (C20H20O10S) showed the greatest docking score with 6lu7. GLU107A, PRO108A, PRO108A, GLN110A, PRO132A, PHE134A, THR198A, MET235A, GLU240A, PRO241A, and THR243A of the amino acid residues interacted with the ligand atom numbers 3053, 3034, 3032, 3053, 3038, 3032, 3046, 3054, 3040, 3033 and 3031 of the ligand with the bond distance of 2.37, 3.40, 3.00, 2.22, 3.78, 4.00, 3.79, 3.54, 3.52, 3.15 and 3.29 (Table S9 and Fig. S7) Jurubine (C33H57NO8) showed optimum docking score with 6lu7. ASN238A of amino acid residues and metal-ligand 2Z and 2Z interact with the ligand atom numbers 3048, 3022, and 3022 of the ligand with the bond distance of 3.18, 2.68, and 2.48 (Table S10 and Fig. S8). Tritriacontan-3-one (C33H66O) showed the best docking score with 6lu7. TYR239A and LUE286A of the amino acid residues interact with the ligand atom numbers 3048 and 3046 of the ligand with the bond distance of 3.89 and 3.60 (Table S11 and Fig. S9). Torvoside F (C45H74O18) showed an adequate docking score of 6lu7. LEU115A, PRO112A, and VAL125A of the amino acid residues interact with the ligand atom numbers 3025, 3038, and 3028 of the ligand with the bond distance of 3.93, 3.47, and 2.88 (Table S12 and Fig. S10). Chlorogenone, Spirostane-3,6-dione (C27H40O4) showed a better docking score with 6lu7. LYS137A, THR199A, LEU286A, LEU286A, LEU287A, and GLU290A of the amino acid residues interact with the ligand atom numbers 3045, 3043, 3048, 3046, 3052, and 3047 of the ligand with the bond distance of 3.46, 3.68, 2.83, 3.82, 3.81 and 3.61 exhibited the best protein-ligand interaction and hydrogen bond distance also showed significant results (Table S13 and Fig. S11).
4. DISCUSSION
The pharmacokinetics properties of natural compounds to be considered as drug candidates were based on Lipinski’s rule of five. This rule is formulated for most orally administered drugs; it uses four criteria to determine if a molecule is drug-like; to have a molecular weight of less than 500, ALogP less than 5 (logarithm of partition coefficient), five or fewer hydrogen bond donor sites, and ten or fewer hydrogen bond acceptor sites. Molecules violating more than one of these rules may have problems with bioavailability. The entire set of compounds well followed the RO5 (Rule of 5) except 15 of the compounds, out of which eight compounds (Alpha carotene, Astaxanthin, Beta carotene, Canthaxanthin, Coenzyme Q10, Epigallocatechin, Gallocatechin, and Theaflavin) violated more than one of these rules and seven compounds violating the only single rule (Chlorogenic acid, Delphinidin, Gallic acid, Lycopene, Myricetin, Vitamin C, Vitamin E). TPSA analysis checked the bioavailability of natural compounds. As per Veber’s rule, for good oral bioavailability, the number of the rotatable bond must be ≤ 10, and TPSA values ≤ 140 Å [32]. The number of rotatable bonds has been shown to be a very good descriptor of oral bioavailability of drugs and has been found better to discriminate between compounds that have an oral bioavailability of drugs. A rotatable bond is defined as any single non-ring bond bounded to a non-terminal heavy (i.e., non-hydrogen) atom. Amide C–N bonds are not considered because of their high rotational energy barrier. The numbers of rotatable bonds in all of our compounds were found to be appropriate as in reference compounds (Donepezil, Galantamine, and Rivastigmine) except four compounds (Vit E, Chicoric acid, CoenzymeQ10, Lycopene) have >10nRB. It was found that only four compounds (Chicoric acid, Chlorogenic acid, Rosmarinic acid, and Theaflavin) have TPSA values ≤ 140Å. The percentage of absorption for all the 45 compounds was calculated using the formula [33]. The Mpro used two experimental structures: Mpro bound to peptide N3 (PDB 6LU7)18 and the unbound form of the protein (PDB 6YB7). The purpose of using two structures for docking is to account to some extent for protein flexibility by receptor ensemble docking [34, 35]. After docking, scoring, and ranking the molecules of the chemical library on each receptor, the hit-lists were merged, and the best ranking pose for each molecule was kept according to the merging-and-shrinking method of Cavasotto and Abagyan [36, 37].
Similar research also showed the reference molecule PRD_002214 of Mpro interacting with several residues and forming three hydrogen bonds with Tyr54, Glu166, and Gln189 and fifteen hydrophobic bonds and yields the binding energy is -7.8 kcal mol-1. Quercetin 3-vicianoside formed hydrogen bonds with His163, Glu166, Ser144, Leu1141, Gly143, and Thr26, while hydrophobic bonds existed with Arg188, Asp187, Met165, His164, Gln189, His41, Thr25, Asn142, Phe140, and Cys145. Absinthin made hydrogen bonds with His163 and hydrophobic bonds with Ser144, Gly143, Cys145, Met49, Met165, Gln189, Pro168, Phe140, Glu166, and Leu14 [38]. The compounds Cholesta-5,7,9-(11)-trien-3-ol,4,4-dimethyl,(3a); Lanosta-7,9-(11),20-triene-3α, 18-diol, diacetate and Cholestan-26-oic acid,3,7,12,24-tetrakis (acetyloxy), methyl ester, (3a,5a,7a,12a) are found to have the affinity of-6.8,-6.9 and-6.9 towards Gr which is very similar to the affinity of the standard (-7.1). These compounds have passed the drug-likeness test. The compounds 9, 12-Octadecadienoic acid, ethyl ester; Hexadecanoic acid, ethyl ester; 9-Octadecenoic acid (Z), methyl ester; Oxacycloheptadec-8-en-2-one, (8Z) have passed the BBB filter. The study revealed that the compounds obtained are therapeutically significant and can be made more potent by further study in the structure-activity relationship of the compounds. This can also be taken as the first step of the research on the antiasthmatic activity of Solanum torvum Sw. [39] (Fig. S12).
CONCLUSION
Most of the PubChem compounds in the fruits of Solanum torvum showed better paramagnetic parameters. The qualitative analysis through this study revealed the presence of the biomolecules such as alkaloids, flavonoids, saponins, tannins, glycosides, and phenolic compounds. Crude extracts and phytochemicals isolated from S. torvum reviewed here have been found to possess antiviral, antioxidant, antifungal, antibacterial, antiulcer, antihypertensive, metabolic-correction, nephroprotective, cardioprotective, antidiabetic, analgesic, anti-inflammatory, immunomodulatory, and erythropoietic activities. Each PubChem compound had unique medicinal properties and can bind to unique binding sites but exerted more pronounced effects than the individual compounds. A combination of twelve compounds gave good results against COVID-19 and can boost the immune system. The compounds showed a good docking score and protein-ligand interaction, which indicated that the compounds have the ability to cure COVID-19 and can act as an effective antiviral agent.
The role of such complex mixtures and the ‘ideal’ composition of an active extract needs to be investigated first using a combination of in vitro (or in vivo animal) techniques in combination with phytochemical or metabolomic techniques. On the basis of biological activities of S. torvum, crude extract and derived phytochemicals and their uses as neutraceuticals and pharmacological agents in traditional and modern research are possible but will first require clinical trials and product development. The current evidence is largely limited to in vitro data.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
Not applicable.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
Not applicable.
FUNDING
None.
CONFLICT OF INTEREST
The authors confirm that this article content has no conflicts of interest.
ACKNOWLEDGEMENTS
Declared none.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s website along with the published article.

