All published articles of this journal are available on ScienceDirect.

In silico Thermodynamic Evaluation of the Effectiveness of RT-LAMP Primers for SARS-CoV-2 Variants Detection
Authors Info & Affiliations
Abstract
Background
Viral mutations are the primary cause of mismatches in primer-target hybridisation, affecting the sensibility of molecular techniques, and potentially leading to detection dropouts. Despite its importance, little is known about the quantitative effect of mismatches in primer-target hybridisation. We have used up-to-date and highly detailed thermodynamic model parameters of DNA mismatches to evaluate the sensibility to variants of SARS-CoV-2 RT-LAMP primers.
Methods
We aligned 18 RT-LAMP primer sets, which underwent clinical validation, to the genomes of the wild-type strain (ws), 7 variants and 4 subvariants, and calculated hybridisation temperatures allowing up to three consecutive mismatches. We calculated the coverage when the mismatched melting temperature fell by more than 5°C in comparison to the matched alignments. If no mismatches were considered, the average coverage found was 94% for ws, falling to the lowest value for Omicron, i.e., 84%.
Results
However, considering mismatches, the coverage was much higher, i.e., 97% (ws) to 88% (Omicron). Stabilizing mismatches (higher melting temperatures) accounted for roughly 1/3 of this increase. The number of primer dropouts increased for each new variant; however, the effect was much less severe if mismatches were considered.
Conclusion
We suggest using melting temperature calculations to continuously assess the trend of primer dropouts.
1. INTRODUCTION
There are eight possible mismatched (MM) base pairs in DNA: AA, AC, AG, CC, CT, GG, GT, and TT. They may arise from DNA replication [1], genetic recombination [2], and primer-template hybridisation in PCR reactions [3], which may lead to false-negative results [4]. Their presence may influence the stability and structural properties of DNA duplex, changing hydrogen bonds conformation and stacking interactions. However, some mismatches show a similar overall shape to a canonical pair and a relatively stable configuration, e.g., a GT-mismatched pair [5, 6]. MM pairs may be found in anti-syn or syn-anti conformations differently from DNA pairs, which are naturally in an anti-anti conformation. Mismatch impact varies from weakly bound (CC pair) to strongly bound (GG pair) in a local conformation, while molecular dynamics and NMR experiments have shown no impact on a global conformation, such as for AA and TT pairs [1, 7]. Internal and terminal mismatches influence primer-target hybridisation in different ways [8]. Mismatches located far from the 3’ end have a moderate effect without influencing PCR performance [3]. On the other hand, those near the 3’ terminal are critical and may lead to non-amplification of the target [8, 9]. Nevertheless, mismatches either near or at the 3’ terminal may avoid false priming unlike the 5’ terminal and internal mismatches [10]. Although it is known that mismatches typically destabilise the primer-target duplex, some types of mismatches are more stable than others, and some even more than AT base pairs, which may contribute towards the stability of the duplex [1, 11, 12]. A few mismatches in PCR primers may contribute to the design of antisense oligonucleotides [13], SNP [14], and allele-specific identification [12].
The isothermal PCR known as RT-LAMP (reverse transcription loop-mediated isothermal amplification) is a robust, fast, and inexpensive molecular technique, and can be carried out in less than an hour [15, 16]. It has been used as a molecular diagnostic test for several diseases, such as ebola [17], zika [18], HIV [19], SARS [20], MERS-CoV [21], and SARS-CoV-2 [22], the causative agent of COVID-19. To detect these diseases, it is necessary to design specific primers to identify the target agent. Unlike PCR, which usually uses a single pair of primers, RT-LAMP uses 2 or 3 pairs: F3 and B3 (outer primers), FIP and BIP (inner primers), and LF and LB (loop primers). The outer and inner primers act at the beginning of the reaction, but just the inner ones act in later cycles. FIP and BIP primers are long primers that contain two parts: F1c and F2 for FIP and B1c and B2 for BIP, which correspond to sense and antisense sequences of the target [23]. Finally, the loop primers are included to accelerate the reaction [24].
For LAMP, in the same way as for PCR [25], mismatches may appear between target and primers due to mutations potentially causing false-negative results [26, 27]. Yet, mismatches may enhance the technique's performance. In fact, SARS-CoV-2 PCR primers designed by Corman et al. [27] during the earlier stages of the pandemic had mismatches that did not hinder the detection of the coronavirus [28]. A few Cas12 enzymes in CRISPR assays have shown mismatch tolerance [29], and resistant mutants may be detected after an antibiotic administration when a mismatch is incorporated at the 3’ terminal [30]. Recent PCR-based methods have used mismatches either at or near the 3’ terminal to detect Delta variant [31] and Omicron subvariants [32].
In a previous study [33], we evaluated the impact of mismatches on RT-PCR primers and probes, where we showed that the mismatches do not always have a negative impact on thermodynamic stability. The reason for this is that there are a number of mismatch configurations that may actually increase the melting temperatures. This was confirmed recently by Scapaticci et al. [34], who found that mutations may have higher melting temperatures and suggested that the melting temperature analysis could be used to detect specific variants. As for PCR, it is expected that mismatches in primer-target hybridisation may appear for LAMP primers, especially for both FIP and BIP primers in which mismatched base pairs in either 5' or 3' ends may prevent the elongation by Bst DNA polymerase, leading to a low amplification efficiency [35]. Although one or two mismatches have been shown to be tolerable for LAMP [30, 36], studies with three or more consecutive mismatches are, to our knowledge, not available.
Here, we have shown the evaluation of DNA mismatches in 18 RT-LAMP primer sets [37-54], which were designed for wild-type SARS-CoV-2 genomes. One of those sets [38] was previously successfully evaluated by us for a few variants and now for amplified genome sets. We applied a previous workflow [33] to analyse those primers for the detection of SARS-CoV-2 variants as Alpha (B.1.1.7), Beta (B.1.351), Gamma (P.1), Delta (B.1.617.2), Lambda (C.37), Mu (B.1.621), and Omicron (B.1.1.529) variants, and BA.2 to BA.5 subvariants. The outcomes show if those primers may still be effective in detecting the variants and how the presence of mismatches may contribute to covering more genomes, consequently detecting the coronavirus. Furthermore, we reinforce the fact that a continuous evaluation of RT-LAMP primer sets is needed to cover variants that may arise, as already suggested [48].
2. MATERIALS AND METHODS
2.1. Genome Sets
We randomly collected 21665 genomes of original SARS-CoV-2 (wild type strain) on 8th October, 2020, at NCBI [55]; 7247 genomes of the Alpha variant, 7497 of the Beta variant, and 2308 of the Gamma variant on 7th April, 2021; 7943 of Delta variant on 5th June, 2021; 7029 of Omicron variant on 16th December, 2021; 6610 of Mu variant, 9340 of Lambda variant, and 7393 and 348 of Omicron subvariants BA.2 and BA.3 on 11th February 2022; and 629 and 1231 of Omicron subvariants BA.4 and BA.5 on 19th September 2022, at GISAID [56].
2.2. Primer Sets
We collected 18 different RT-LAMP primer sets designed for SARS-CoV-2 original genomes that underwent clinical validation [37-54], resulting in a total of 436 primers. Their details are shown in Table S1. FIP and BIP primers were divided in F1c/F2 and B1c/B2 primers, respectively, except those from three sets [38, 40, 51], for which the division of primers was already given. We found all possible combinations of primer pairs and selected those according to the temperatures of the same type of pair from the three sets just mentioned.
2.3. Evaluation Workflow
All primers were aligned to each genome set using a Smith-Waterman algorithm, as described earlier [33]. Fully matched alignments were called strictly matched and those with single, double, and triple consecutive mismatches were termed partially matched. Alignments with four or more consecutive mismatches were considered as not aligned. The limit of the maximal number of consecutive mismatches is be due to the fact that the available parameter only covers up to three contiguous mismatches [11]. In addition, it is very likely that four or more mismatches will destabilize the primers far beyond the limits considered here. Also, deletions in the viral genome, as in the Omicron variant [57, 58], may lead to no alignment of the primers.
Hybridisation temperatures for matched (Tref.) and mismatched (TMM) alignments were calculated from a mesoscopic model with the parameters obtained from a previous work [11].
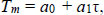 |
(1) |
The reference hybridisation temperature Tref. for each primer is shown in Supplementary Table S1. It should be noted that the parameters [11] are for a sodium buffer, which is different from those typically used in PCR reactions that contain Mg+. Therefore, the absolute temperatures Tref. may be different from the actual melting temperatures of the primers. However, since our analysis deals with temperature differences, which are not strongly buffer-dependent, we expect them to be sufficiently accurate for our purposes.
We define a strictly matched (AT and CG only) alignment coverage for each primer as
follows:
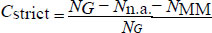 |
(2) |
Where, NG is the total number of genomes, Nn.a. is the number of genomes for which no alignment was found, and NMM is the number of genomes for which a partial alignment containing mismatches was found.
The difference between reference hybridisation temperature Tref. and mismatched alignments TMM is defined as follows:
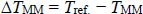 |
(3) |
Where, TMM is usually lower than Tref. [11]. The partial coverage for alignments with up to three contiguous mismatches is defined below:
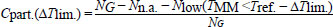 |
(4) |
Where, Nlow is the number of alignments where the mismatched melting temperature TMM is lower by ∆Tlim. than the reference Tref. It should be noted that as there are many mismatch configurations that have an increased TMM, that is, there are situations where Cpart. > Cstrict even for ∆Tlim. = 0. A previous work has provided additional details of this workflow [33].
All 18 primer sets were aligned against the genomes of SARS-CoV-2 variants. We calculated the hybridisation temperatures and coverages for both matched and mismatched alignments considering single, double, and triple consecutive mismatches. The complete evaluation was carried out in approximately 120 h computing time.
2.4. Availability
The software packages used to carry out this work are freely available and can be found at https://bioinf.fisica.uf mg.br/software/, in the analyse primer lamp.tar.gz package.
3. RESULTS AND DISCUSSION
We assessed 18 clinical validated RT-LAMP primer sets [37-54]. They showed high strict and partial coverages for wild-type SARS-CoV-2 and its variants. Even for variants and subvariants, a few primers achieved more than 90% coverage. A considerable number of primers showed high coverages only when mismatches were taken into account. Also, primers utilized by Alves et al. [38] achieved high coverages for wild-type SARS-CoV-2 and Gamma variant, in agreement with the experimental results. Furthermore, Almeida et al. [59] showed E1 and N2 primer subsets to be able to identify the Omicron variant target despite the presence of only a single mismatch. All strict and partial coverages are shown in Tables S2-S55.
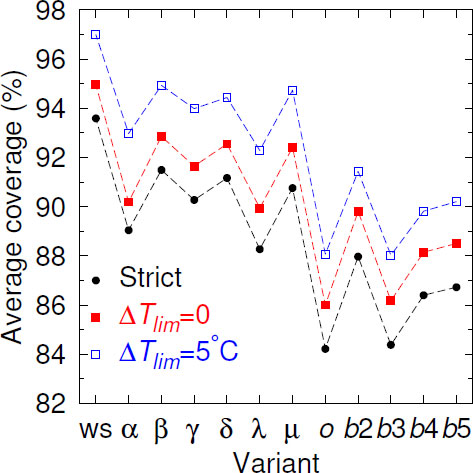
Given the continuous mutation of the SARS-CoV-2 genomes, it is expected that over time, mismatches should increasingly occur within the primer regions. Fig. (1), where we show the Cstrict averaged over all 436 primers, illustrates this decreasing coverage as variants appear. In comparison to the wild-type strain (ws) coverage, all variants decreased their coverage. When we considered partial coverages in the presence of mismatches with ∆Tlim = 0°C, that is, primers with TMM ≥ Tref., the curve uniformly shifted upwards. For ∆Tlim = 5°C, Beta, Gamma, Delta, and Mu partial coverage became slightly higher than the ws strict coverage. However, the rate of decrease was not uniform, and some variants had higher coverage than their presumed predecessor variants. For the Omicron variant, which had a larger number of mutations [60], we have observed a sharp drop in the coverage. However, for the subsequent subvariant, the picture has been mixed; the BA.3 subvariant shared the low coverage, but BA.2, BA.4, and BA.5 have shown a higher coverage. The reason for this oscillation was not clear.
| Cpart.(0°C) (%) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Primer | ws | Alpha | Beta | Gamma | Delta | Lambda | Mu | Omicron | BA.2 | BA.3 | BA.4 | BA.5 |
| As1e F1c [28] | 99.2 | 99.4 | 98.7 | 99.7 | 99.2 | 99.0 | 99.7 | 98.5 | 100 | 98.6 | 99.5 | 99.7 |
| iLACO-F1c [28] | 99.2 | - | 99.1 | 99.5 | 99.3 | 99.8 | 99.8 | 99.5 | 99.8 | 99.7 | 100 | 99.9 |
| N15-B1c [33] | 97.7 | 98.8 | 96.5 | 98.4 | 99.9 | 99.4 | 99.6 | 99.2 | 99.9 | 99.7 | 98.9 | 98.9 |
| N1-B1c [35] | 98.4 | 99.5 | 97.1 | 99.6 | 99.6 | 99.7 | 99.3 | 93.8 | 99.8 | 94.3 | 99.0 | 98.7 |
| N1-F1c [35] | - | - | - | - | - | - | - | 92.7 | 97.5 | 94.0 | - | - |
| N2-F1c [35] | 99.1 | 99.1 | 96.6 | 99.1 | 99.9 | 98.7 | 99.7 | 99.3 | 99.9 | 99.7 | 99.0 | 99.0 |
| NEB orf1a-A-F1c [37] | 98.9 | 99.5 | 99.2 | 99.5 | 99.8 | 99.6 | 99.1 | 99.4 | 99.7 | 99.1 | 99.2 | 96.9 |
| F1c [38] | - | - | - | - | - | 98.9 | 99.2 | 93.9 | 99.5 | 93.1 | - | - |
Similar to what we have seen for RT-PCR [61], many alignments that would result in a null strict coverage achieve partial coverage beyond 99% if mismatches are considered. In some cases, a large partial coverage is already obtained for ∆Tlim = 0°C, that is, if we consider only mismatches that do not destabilize the duplex.
In Table 1 we show a few examples of primers that have zero strict coverage but go beyond 90% if stabilizing mismatches are considered. It is somewhat surprising that some primers achieved high coverages for Omicron only and not for the other variants, despite the fact that all were designed for the wild-type strain. While this seems to be an opposite trend to the overall decline for Omicron, one should note that a higher coverage for Omicron was rather exceptional and only occurred for very few primers. On the other hand, this quite clearly highlights that the assessment of mismatch influence is far from trivial.
| Ref. | Nprimers | Ndrop | ws | Alpha | Beta | Gamma | Delta | Lambda | Mu | Omicron | BA.2 | BA.3 | BA.4 | BA.5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| [29] | 32 | 6 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 3 | 3 | 3 | 4 | 3 |
| [30] | 40 | 7 | 0 | 1 | 1 | 1 | 1 | 0 | 0 | 3 | 3 | 3 | 3 | 3 |
| [31] | 81 | 28 | 4 | 11 | 5 | 5 | 5 | 6 | 12 | 9 | 5 | 10 | 7 | 6 |
| [32] | 8 | 1 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 |
| [33] | 32 | 6 | 0 | 0 | 0 | 1 | 0 | 3 | 1 | 3 | 2 | 3 | 2 | 2 |
| [34] | 15 | 3 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 2 | 2 | 2 | 2 |
| [35] | 32 | 5 | 1 | 3 | 1 | 3 | 2 | 2 | 1 | 2 | 2 | 2 | 3 | 2 |
| [36] | 8 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1 | 1 | 1 | 1 |
| [37] | 32 | 5 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 1 | 2 | 2 | 4 | 2 |
| [38] | 10 | 6 | 0 | 2 | 1 | 0 | 2 | 3 | 0 | 3 | 3 | 3 | 3 | 3 |
| [39] | 48 | 12 | 0 | 3 | 0 | 5 | 3 | 6 | 0 | 7 | 7 | 7 | 8 | 7 |
| [40] | 8 | 5 | 0 | 2 | 0 | 1 | 0 | 0 | 2 | 0 | 0 | 0 | 0 | 0 |
| [41] | 16 | 5 | 0 | 1 | 2 | 0 | 1 | 1 | 0 | 1 | 1 | 1 | 1 | 1 |
| [42] | 8 | 2 | 1 | 1 | 1 | 2 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| [43] | 15 | 3 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 1 | 1 | 1 | 1 | 1 |
| [44] | 15 | 2 | 0 | 1 | 0 | 1 | 1 | 2 | 0 | 1 | 1 | 1 | 1 | 1 |
| [45] | 16 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 | 1 |
| Primers | Mismatches | Tref. (°C) | TMM (°C) |
|---|---|---|---|
| N1-B1c | GT | 73.9 | 80.6 |
| N2-B1c | TT/CT | 69.0 | 66.6 |
| ORFlab-1-F1c | GT/TT | 71.0 | 70.1 |
| N2-F1c | GT/CT/TT | 75.6 | 79.9 |
While most primers had large coverages, an important amount of primers failed to achieve significant coverage for at least one variant, and may represent a potential dropout. A summary of the amount of primers that could potentially represent dropouts is provided in Table 2. Here, we have considered a very stringent threshold of 5% at ∆Tlim = 5°C, that is, primers where even considering a maximal 5°C melting temperature below the reference temperature covered less than 5% of the available genomes for a given variant. Only the set proposed by Alekseenko et al. [37] had no potential dropout primers at all. The complete list of potential dropout primers for each variant is shown in Tables S56-S67.
Mismatched pairs in 5' or 3' terminals of FIP and BIP primers may hamper the amplification by Bst DNA polymerase. However, we found a few alignments with either 5' and 3' terminal mismatches to have a hybridisation temperature within the threshold and contribute to the increase in the coverage when mismatches are taken into account. Clearly, in some cases, mismatches in both terminals reduced the temperature. An interesting case was found for four primers from the work of Ji et al. [44], which showed single, double, and triple contiguous mismatches at the 3’ terminal (Table 3). We observed that only the double mismatched pair cases decreased the temperature. On the other hand, the single and triple mismatched pairs increased the temperature. Perhaps, due to the GT mismatched pair has been reported as a strong pair [2, 11, 62, 63]. FIP and BIP primers with terminal mismatches that had an increase in their coverage are shown in Tables S68-S79 for each genome set. It should be noted that FIP and BIP primers were divided into F1c/F2 and B1c/B2, respectively, and as such treated individually. With respect to the LAMP technique, the F1c and B1c depend on their respective F2 and B2 complements, and the dropout may in practice be higher.
CONCLUSION
In this work, we have evaluated the coverage of 18 RT-LAMP primer sets considering single, double, and triple mismatches in primer-target hybridisation to SARS-CoV-2 variants. In general, the average coverage of these primer sets decreased for the new variants, when compared to the wild-type strain. Overall, the coverage was lowest for the Omicron and BA.3 variants. However, a clear monotonic decrease in the coverage was not observed; instead, for some variants, the coverage increased when compared to its putative predecessor, as exemplified most notably by the Mu variant, which showed one of the highest coverages. Coverage uniformly increased if mismatches were taken into account, while not enough to completely compensate for the loss in comparison to the wild-type strain, as is shifted the worst case from 84% to 88%. Similarly, the number of potential dropout primers increased with each new variant, and only one out of 18 sets showed no potential primer drop-out. We suggest the use of the methodology described here to continuously evaluate the effectiveness of RT-LAMP primer as new variants emerge. Furthermore, our method can be applied to the detection of other infectious diseases.
LIST OF ABBREVIATIONS
| α | = Alpha |
| β | = Beta |
| γ | = Gamma |
| δ | = Delta |
| λ | = Lambda |
| µ | = Mu |
| o | = Omicron |
| b2 | = BA.2 |
| b3 | = BA.3 |
| b4 | = BA.4 |
| b5 | = BA.5 |
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No humans/animals were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data and supportive information are available within the article.
FUNDING
PM is supported by Coordenação de Aperfeiçoamento de Pessoal de Nı́vel Superior (Capes/Ação Emergencial, Brazil, Finance Code 001). PA is supported by the Brazilian Ministry of Science, Technology and Innovation, through the “Rede Virus” (MCTI, FINEP grant number 01.2N5.00). RMN is supported by Fundação de Amparo à Pesquisa do Estado de Minas Gerais (Fapemig grant number PPM-00699-18). RMN and GW are research fellows from Conselho Nacional de Desenvolvimento Cientı́fico e Tecnológico (CNPq grant numbers RMN 312965/2020-6, GW 307538/2019-2).
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.
SUPPLEMENTARY MATERIAL
Table S1 shows all primers used and their reference temperatures. Tables S2-S55 show the strict and partial coverages of all primers. Tables S56-S67 show the potential drop-out primers for each variant. Tables S68-S79 show the terminal mismatched positions of FIP and BIP primers for each genome.