All published articles of this journal are available on ScienceDirect.

SARS-CoV-2, Inflammatory Apoptosis, and Cytokine Storm Syndrome
Authors Info & Affiliations
Abstract
SARS-CoV-2 (Severe Acute Respiratory Syndrome Coronavirus 2), a novel and currently intensively studied beta coronavirus, is the causing agent of COVID-19 (Coronavirus Disease 2019), a highly contagious and devastating disease that has killed more than 2 million human beings since December 2019. Building on what has already been understood from studying SARS-CoV, a closely related single-strand RNA virus that set off SARS in 2002 and 2003, researchers began to learn how SARS-CoV-2 operates its vicious effects on the host cells. In essence, COVID-19 patients display hyperinflammatory and dysregulated cell death phenotypes that give a spectrum of symptoms ranging from mild to moderate upper-respiratory tract illnesses. However, SARS-CoV-2 can elicit serious pathologies, such as acute respiratory distress syndrome, sepsis-like multi-organ failure and even death, depending on the individual and their pre-existing condition(s). As viruses cannot reproduce independently, they hijack the machinery within the host cells and enslave them for the purpose of propagation. SARS-CoV-2 RNA genome harbors the genes that produce the protein products for manipulating host cell, viral replication, and repeating the vicious viral cycle. For counteracting the viral invasion, human cells have developed layers of defense mechanisms, such as restriction factors, Regulated Cell Death (RCD) pathways, interferon production, inflammatory response, and innate and adaptive immunity that are used to recognize and thwart viral infection. Unfortunately, some coronavirus encoded proteins are capable of attacking the host anti-viral system to achieve parasitic advantages. We reviewed the proteins of SARS-CoV and SARS-CoV-2 that possess manipulating effects on the host cell and cause tissue damage, immune cascade, cytokine production and release. We also discuss the means to restore the homeostatic balance between inflammatory response and RCD pathways and the potential targeted interventions that can be used to treat and/or prevent COVID-19.
1. INTRODUCTION
1.1. SARS-CoV and SARS-CoV-2
Coronaviruses (CoVs) are a diverse group of positive-sense single-strand RNA viruses, named for their distinctive spiked envelopes. CoVs infect many different species, with α and β types, commonly infecting humans [1, 2]. Most CoVs infections are asymptomatic or induce mild upper respiratory symptoms, as in the common cold. However, in the last two decades, more virulent strains of CoV have emerged, which cause lower respiratory symptoms with complications leading to significant mortality. In 2002, Severe Acute Respiratory Syndrome Coronavirus (SARS-CoV) emerged, infecting > 8,400 people worldwide and killing > 900. A decade later, Middle East Respiratory Syndrome Coronavirus (MERS-CoV) emerged, infecting > 1,400 and killing 39% of those infected [3]. Currently, SARS-CoV-2, the causing agent of COVID-19, is sweeping the globe, killing more than 2 million people with more than 96 million known infections so far (as of January 20, 2021; https://coronavirus.jhu.edu/map.html). After intensive studies for several months, it is now known that SARS-CoV-2 can cause various degrees of severity of COVID-19 disease, ranging from asymptomatic to life-threatening [4, 5]. The most severe cases are associated with virally driven hyperinflammation, downregulation of type 1 interferons (IFN-I), cytokine storm syndromes, and sepsis-like multi-organ failure [2, 6]. With the periodic emergence of such pathogens, researchers are working furiously and collaboratively to learn more about this virus, building on what has already been learned from studying SARS-CoV, MERS-CoV and other CoVs. It is pivotal to develop prevention, diagnosis, and treatment strategies for the present danger and prepare for future outbreaks.
2. PROTEINS ENCODED BY SARS-CoV AND SARS-CoV-2
Viral infections have a central goal: reproduction. As viruses cannot reproduce independently, they entirely rely on coopting functioning cells to hijack the machinery within the host cells and enslave them for the purpose of propagation and then the tactic is the same, i.e., infiltrate, reproduce, and spread. Thus, viral genomes, produce protein products for infection, manipulating host cells replicating, and repeating the vicious viral cycle. SARS-CoV is a fully enveloped RNA virus with a genome of 29,751 bases which contains a number of Open Reading Frames (ORFs) that encode several enzymes necessary for protein processing, genome replication, viral transcription, and translation [7]. The RNA genome also encodes for the structural proteins, namely, Spike Protein (S), Envelope Protein (E), Membrane protein (M), Nucleocapsid protein (N), sixteen Nonstructural proteins (Nsps), and 15 so-called Accessory proteins (APs, also known as ORFs) [1-3, 8, 9]. Based on the sequence homology analysis, SARS-CoV-2 and SARS-CoV share about 80% sequence identity at the amino acid level, suggesting a high degree of functional conservation [9-11]. Using SARS-CoV as an example, the 5’ end of the genome begins with an untranslated region, followed by the first ORF in two overlapping sections, a and b [7, 12]. Comprising two-thirds of the genome, ORF1a and 1b encode for polyproteins which, after processing and modifications, generate 16 Nsps. Some Nsps are known to interact with host cell function, for example, in viral genome’s replication and translation [8, 9, 12].
Nsp1 has been shown to suppress host expression of mRNA, including IFN-I, which would otherwise begin a pro-inflammatory host response [13]. Nsp3 is known to stifle INFs, cytokine release, IRF3 and NF-κB. Interestingly, SARS-CoV-2 Nsp3 is shown to have a PLPro domain, which cleaves the polyproteins at specific sites [3, 9, 12]. Nsp5 of SARS-CoV-2 also contains a protease in the form of a 3CLpro domain, where as Nsp 6 inhibits IRF3, blocking the production of IFN-I [3, 12]. SARS-CoV-2 Nsp 7 and 8, together form a primase and Nsp9 has nucleic acid binding activity [14]. Recently, Viswanathan et al. resolved the structure and function of the heterodimer of Nsp16/10 for SARS-CoV-2, [15]. SARS-CoV-2 Nsp12 is likely to be an RNA-dependent RNA polymerase, Nsp13, a helicase, Nsp14, an endonuclease and Nsp15, a Poly(U)-specific endoribonuclease [12]. S protein of both SARS-CoV and SARS-CoV-2 are trimers involved in the host cell entry, initiating cell-cell fusions [1, 10]. It uses host membrane receptor ACE2 and protease TMPRSS2 to enter a cell. The cleavage product from the S protein can interact with membrane Toll-like Receptors (TLRs), setting off an immune cascade, or acting as a decoy for extracellular immune action, preventing the marking, or it can capture the virion while it gains entry.
2.1. Proteins of SARS-CoV/SARS-CoV-2 in Regulating Inflammatory Apoptosis
For counteracting the viral invasion, human cells have formed defense systems that recognize and attempt to obstruct viral infection. There are several layers to this host defense system, specifically, viral restriction factors, RCD pathways, inflammatory response, interferon production, and innate and adaptive immunity. The tight spatial-temporal regulation of the crosstalk between these pathways is the key in halting or impeding viral infection and spread. If unfortunately, viruses are successful in their invasion, a few infected cells sacrificing themselves via RCD to diminish the viral replication and to be able to reproduce is crucial. Thus, some SARS-CoV proteins have developed the function to manipulate host cell RCD pathways, in particular, apoptosis [9, 11, 16-18].
Apoptosis was the first characterized form of programmed and regulated cell death [19, 20], which proceeds in a physiologically and biochemically orchestrated manner to execute cell suicide. It has been well documented that apoptosis tightly regulates growth and differentiation, protects against disease progression, and thwarts microorganism infection. The functions by which the apoptotic program can be activated are tightly regulated through the activation and inhibition mechanisms within the cell, controlled through gene products, post-translational modifications, sequestration of key components, or zymogens (such as pro-caspases) awaiting death signals to mobilize them into action. On a molecular basis, apoptosis is characterized by the activation of caspase and the release of pro-apoptotic factors from the mitochondria and other organelles. Morphologically, apoptosis is characterized by cell shrinkage, chromatin condensation, ROS generation, and membrane blebbing [20-23]. Apoptosis serves the essential purpose of ensuring tissue and organismal homeostasis via two distinct pathways: intrinsic and extrinsic. The intrinsic pathway is initiated from internal signals, such as DNA damage and oxidative stress and the extrinsic pathway is induced by a variety of factors, such as viral infection. Apoptosis is a powerful and the most efficient host defense mechanism in virally infected cells that halts the viral progression and keep the infection from getting out of control. Some viruses have evolved to manipulate the extrinsic pathway of apoptosis in order to ensure host damage and weakening of defenses. The extrinsic pathway of apoptosis can be mediated by death receptors, such as TNFR and TRAIL on the host cell surface and is exploited by viruses as a mechanism to disseminate (Fig. 1). The exploitation of the apoptotic pathway by SARS-CoV-2 appears to have been co-opted by this viral strain such that apoptosis and its function in the innate immune response to halt or slow viral infection progression and spread is modulated to maximize the spread of SARS-CoV-2 [9, 24]. The apoptotic program in regard to immune defense mechanisms is evolved to remove cells that have become virally infected from participating in the viral reproduction program, whereby it orchestrates destroying the cell and its constituent machinery. Apoptosis, if “leaky”, may allow for some viral partials to spread, but a tightly controlled apoptosis reduces overall viral titer and inhibits the potential amount of virus that can be released to infect other neighboring cells within an organism [25]. Apoptosis represents an ordered system of events that orchestrate the final fate of a cell, such that a single cell can systematically be removed.
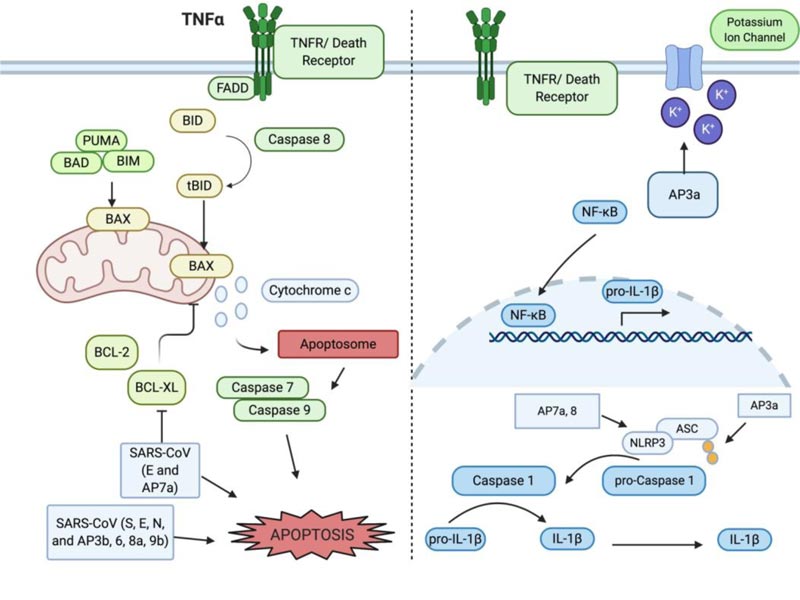
Inflammation is usually the very first response when host cells encounter viral infection that, in part, induces apoptosis that is largely mediated by the proinflammatory cytokines, such as interleukin 1β (IL-1β). IL-1β is synthesized as the inactive form pro- IL-1β and activated via proteolytic cleavage. Cleavage of pro- IL-1β into the active form is mediated by caspase 1. Caspase 1, which is also synthesized in the inactive form pro-caspase 1, is activated through the NLRP3 inflammasome [26, 27]. The NLRP3 inflammasome is formed in the presence of a pathogen-associated molecular pattern (PAMP, for example, LPS) being recognized by Pattern Recognition Receptors (PRR, for example, toll-like receptors and nod-like receptors) on the host cell. The adaptor protein ASC provides a link between the inflammasome and pro-caspase-1 to cleave and activate it. Many other regulators and signaling molecules can dictate the outcome of apoptosis following a viral infection. One of these players is TNF receptor-associated factor 3 (TRAF3), whose signaling is normally associated with the induction of the NF-κB pathway, prompting transcription of several pro-inflammatory cytokines. Changes in the ubiquitination of TRAF3 can change the outcome of NF-κB activation and IFN induction [28]. In the context of SARS-CoV, TRAF3 mediates ubiquitination of p105 and ASC in order to trigger the activation of pro-IL-1β gene transcription [29].
SARS-CoV-2 patients and particularly those in serious condition have a highly increased level of circulating cytokines, for example, IL-2, IL-6, IL-7, Granulocyte Colony-stimulating Factor (GM-CSF), interferon-γ inducible protein 10 (IP-10), Monocyte Chemoattractant Protein 1 (MCP-1 or CCL2, a C-C motif chemokine ligand-2), and Macrophage Inflammatory Protein 1-α (MIP-1α or CCL3) [6, 27, 30-32]. This so-called Cytokine Storm Syndrome (CSS) can be due to the dysregulated response to viral infection. One mechanism by which cytokines are upregulated involves the recognition of danger signals by Nod-like Receptors (NLRs), resulting in the activation of caspase 1, which can trigger hyperinflammatory cell death, for example, pyroptosis or ferroptosis [33, 34]. Caspases serve a variety of functions in apoptosis as well, operating as either initiators or effectors of cell death.
Infection of SARS-CoV is determined by the binding of the S protein to host cell surface receptor angiotensin-converting enzyme 2 (ACE2) and the serine protease TMPRSS2 for S protein priming [10, 35]. Studies in SARS-CoV found the induction of massive apoptosis in the respiratory tract, the thyroid gland, and the liver [36]. This process is thought to be via the upregulation of the pro-apoptotic Bcl-2 family member, Bax. Interestingly, SARS-CoV replication was only mildly impacted by caspase inhibition. The Bcl-2 family of proteins play diverse and critical roles in the execution of apoptosis. The family can be divided into pro-apoptotic, including BAX and BAK, anti-apoptotic, including Bcl-2 and Bcl-XL, and BH3, such as PUMA, Noxa, and ApoL6 [20, 37, 38]. These proteins can dictate if a cell survives or proceeds through apoptosis. Interactions between viral proteins and the Bcl-2 family can manipulate the decision between survival and apoptosis. SARS-CoV infected cells have demonstrated apoptotic phenotypes, such as cell shrinkage and membrane blebbing. Cell death in these cases was found to occur in a viral-replication and time-dependent manner, involving the downregulation of the anti-apoptotic Bcl-2, upregulation of the pro-apoptotic BAX and the activation of effector caspase 3. The upregulation of Bax indicates that the mitochondrial and intrinsic pathway is involved in SARS-CoV-induced apoptosis (Fig. 1). Thus, central to the apoptotic program are distinct regulation mechanisms that represent prime targets for manipulating or definitively controlling the apoptotic program. The Bcl-2 family of proteins balances the relative pro- and anti-apoptotic outcomes [39]. SARS-CoV has been demonstrated to use some AP proteins to control host cell death. For example, AP7a targets and co-localizes with Bcl-XL in the endoplasmic reticulum and mitochondrial membrane and likely inhibits the pro-survival function of Bcl-XL (Table 1) Furthermore, direct manipulation of Bcl-2 proteins and caspase activity within the cell can also modulate apoptosis, which has been shown in Vero cells infected by SARS-CoV [17, 40]. Thus, SAR-CoV proteins can manipulate host cell apoptosis in favor of fine tuning viral replication and spreading as well as avoiding host innate immune responses [33]. Similarly, it has been shown that MERS-CoV activates both the extrinsic and intrinsic pathways to facilitate the apoptotic cell death program and being indicative of the significant damage to the renal organ system due to large-scale induced apoptosis [41, 42]. Thus, invading viruses can manipulate host cell apoptosis in 2 different ways: at the initial phase of viral infection, some viral proteins inhibit apoptosis so that the host cell is enslaved for producing more viral particles. At the later stage of viral infection, some viral proteins induce noncanonical apoptosis for releasing viral particles from the dying cell [10, 20, 23]. The known functions of accessory proteins (APs) that regulate apoptosis are summarized below [8, 17, 43].
2.2. AP3, AP3a, and AP3b
The 3’ end of the SARS-CoV genome harbors 15 major open reading frames, seven of those encoding for the Accessory Proteins (AP) 3a, 3b, 6, 7a, 7b, 8, 9b, 9c, and 10. AP3 is split into 3a and 3b for SARS-CoV, however, in the new SARS-CoV-2, it remains as one protein. AP3a and 3b of SARS-CoV are expressed on the viral surface and can be used as markers in the majority of SARS-CoV patients, however their structure is yet to be determined. Interestingly, AP3a is detectable in host cell cytosol in a distinct punctate pattern, as well as in the plasma membrane. AP3a contains a cysteine-rich domain that enables the protein to dimerize and form potassium permeable ion channels. Blocking of these ion channels can suppress viral-mediated apoptosis, pointing to a role in apoptosis for AP3a (Table 1) [14]. Induction of apoptosis in the presence of AP3a overexpression in Vero E6 cells was determined by DNA fragmentation and chromatin condensation [44, 45]. in vitro studies also showed that AP3a interacts with AP7a, M, S, and E. It can be detected in SARS patients and is co-localized with the M and E proteins, indicating that AP3a is critical for the function and assembly of SARS-CoV. Specifically, the tyrosine-based sorting motif ΔYXXΦ of AP3a was found to be essential for transport from the Golgi to the plasma membrane. If this transport is blocked, AP3a is degraded in the lysosome [46]. Recently, the role of AP3a in apoptosis in SARS-CoV led to the investigation of a role for AP3 in apoptosis in the context of SARS-CoV-2. Ren et al. showed that AP3a of SARS-CoV-2 can efficiently induce apoptosis in various cell lines, including Vero E6, HEK293T and HepG2 [47]. However, when comparing the pro-apoptotic activity, AP3 of SARS-CoV-2 has a relatively lower killing activity than that of AP3a of SARS-CoV. Expression of SARS-CoV AP3a was found to induce NF-κB activation and increased production of cytokines, resulting in cell death [29]. AP3a can induce the cytokine storm via the activation of the NLRP3 inflammasome and protein maturation of pro- IL-1β, as well as TRAF3-dependent ubiquitination of p105 and ASC. In addition, AP3a can cause upregulation of fibrinogen, further contributing to the increased cytokine storm [27, 29]. Thus, additional research to examine the role of AP3a in SARS-CoV-2 infection is needed. Positive selection of 3a along with AP8 has been suggested to drive the early divergence events of the pandemic [48].
| SARS-CoV Protein | Function | References |
|---|---|---|
| AP3a | Induce NF-κB pathway activation | [27] |
| Upregulation of IL-1β and other pro-inflammatory cytokines | [14, 44] | |
| Role in the cytokine storm | [47] | |
| Forms membrane ion channels | [29, 45] | |
| AP3b | Blocks host cell cycle at the G0/G1 | [48, 50] |
| Possibly induce necrosis | [49] | |
| AP6 | Interferon antagonist | [8, 60] |
| Induces apoptosis via a caspase 3 dependent mechanism | [52-55] | |
| AP7a | Blocks host cell cycle at the G0/G1 phase | [56] |
| Triggers apoptosis via interactions with the pro-apoptotic Bcl-XL | [17, 43] | |
| Activates the transcription factor NF-κB | [57] | |
| AP8 | Induction of apoptosis via caspase 3 | [58] |
| AP9 | Transport in and out of the nucleus may upregulate apoptosis | [41, 59, 61-63] |
Although, less extensively studied than AP3a, AP3b also has a role in cell death. It has been shown that AP3b induces apoptosis via cell cycle arrest at the G0/G1 phase [49]. Another study indicated that AP3b can induce necrosis in Vero E6 cells and has also been detected in the tissues of SARS-CoV patients [50]. Notably, AP3b has been reported to be missing in many SARS-CoV-2 cases, so it may not play as significant of a role in COVID-19 infection [51].
2.3. AP6
AP6 is a 63 amino-acid membrane protein that is incorporated into mature virions and associated with the Golgi apparatus and endoplasmic reticulum. Previous studies have shown an increase in virulence in mice that overexpressed AP6, suggesting that AP6 may play a critical role in the association with structural proteins that require viral recruitment, replication, and pathogenesis (Table 1) [8, 52]. AP6 contains a highly polar C-terminal region and an amphipathic N-terminal region associated with its enhanced viral capabilities [53]. AP6 was also found to be an interferon antagonist, inhibiting IFN-β synthesis and signaling, suggesting the role of AP6, not only for enhancing viral replication but also for inducing apoptotic cell death. The induction of apoptosis occurs in a caspase-3 dependent manner in the presence of AP6 and AP7a. It was also confirmed that AP6 induced cell death can be blocked by caspase inhibitor, z-DEVD, as well as a JNK inhibitor. In addition, AP6 was shown to co-localize with Nsp8 in SARS-CoV infected cells, further demonstrating a role in viral replication [54, 55].
2.4. AP7a
Among all the alpha and beta coronaviruses, only SARS-CoV and SARS-CoV-2 contain the AP7a. Before the discovery of SARS-CoV-2 that caused the COVID-19 pandemic, SARS-CoV was the only coronavirus known to possess the AP7a. This fact led to the investigation of the role of AP7a in the widespread cell death that was seen in SARS patients. AP7a was found to induce apoptosis in a caspase-dependent manner (Table 1). Tan et al. showed that AP7a-induced apoptosis occurs upstream of the Bcl-2 family as indicated by the capability that pro-survival Bcl-XL blocks AP7a-induced apoptosis in SARS-CoV infected cells [40, 43]. This suggests that apoptosis triggered by AP7a occurs via the direct interference/binding of Bcl-XL. AP7a is critical for viral-induced apoptosis, however, it has been shown that AP7a is dispensable for viral replication [56]. In addition, AP7a has been shown to block host cell cycle progression at G0/G1 phase via the cyclin D3/pRb pathway [57].
2.5. AP8
AP8 has also been shown to induce apoptosis via a caspase 3 dependent pathway in vitro (Table 1) [58], however, more studies are warranted in the understanding of AP8’s role in vivo.
2.6. AP9
AP9b is 98 amino-acid lipid and membrane binding protein that has been suggested a role in the viral assembly of SARS-CoV [59]. In SARS-CoV, AP9b has been shown to co-precipitate and interact with AP6 in SARS-CoV infected cells (Table 1) [60]. Both proteins were found to partially co-localize to the cytoplasm. This protein-protein interaction could be contributing to the role of AP6 in viral replication and induction of apoptosis. However, more work needs to be done to determine if these interactions are maintained in the new SARS-CoV-2 and what impact, if any, these interactions may have on the RCD of host cells and the progression of COVID-19. Importantly, AP9b has been shown to enter the nucleus, either via nuclear localization signal or passive transport and can then interact with the Crm1 protein to be exported out of the nucleus. This interaction was shown to be essential to properly degrade AP9b and the blocking of this interaction, leading to an increase in apoptosis [61].
Recently, AP9b of SARS-Cov-2 was found to share 70% sequence homology to the counterpart of SARS-CoV. The two proteins have minor differences in their structure, such as an increased number of strands and helices in SARS-CoV-2 [62]. Moreover, antibodies to AP9b were detectable in the sera of SARS-CoV patients, suggesting that it plays an important role in SARS-CoV infection.
The C-terminal subunit S2 has been found to be capable of inducing intrinsic apoptosis via the upregulation of cytochrome c and down-regulation of pro-survival members of the Bcl-2 family in the viral susceptible Vero E6 cell line [63]. Similar to AP3, S2 has been suggested to affect cell cycle arrest at the G1/S phase [41].
During SARS-CoV infection, pro-apoptotic or anti-apoptotic pathways of host cells seem to be altered in various stages, suggesting that SARS-CoV proteins may be regulated spatially and temporally at different stages of viral progression [43]. Interestingly, SARS-CoV-induced apoptosis can be partially reversed via the expression of Bcl-2, suggesting that apoptosis is primarily mediated by Bcl-2 and caspase- dependent-pathway [64]. Overall, most of the apoptosis-regulating proteins in SARS-CoV are present in SARS-CoV-2, so more research should be done, to better understand the pathophysiology and potential treatment of COVID-19.
In addition to AP proteins, other SARS-CoV proteins have been shown to possess cell death activities. For example, M protein facilitates viral assembly and activates both caspase 8 and 9, triggering apoptosis [8, 40, 65], whereas N protein can function as a suppressor of RNA sensing, to interfere with host defense by blocking IFN-I [1, 2]. Interestingly, SARS-CoV E protein has been shown to trigger apoptosis in T cells through a BH3-like domain at its carboxyl-terminal that interacts with Bcl-XL [66]. Interestingly, E protein can function as an ion channel, similar to protein AP3a, and has been found to promote activation of the NLRP3 inflammasome, stimulate the NF-kB pathway, and increase the production of IL-1β [67]. E protein of SARS-CoV-2 shares 95% sequence homology with that of SARS-CoV [9] at the protein sequence level, suggesting functional conservation of E protein in SARS-CoV viruses. Thus, blocking E protein could limit cytokine storm, inflammasome activation and dysregulated apoptosis [68, 69]. Recently, some small compounds, for example, HMA (5-N,N-Hexamethylene amiloride), BIT225, and AMT (1 amino-adamantane), have been shown to block E protein of SARS-CoV [68]. Gupta et al. found three phytochemicals, Belachinal, Macaflavanone E, and Vibsanol B, isolated from the species Belamcanda chinensis, Macaranga tanarius and Viburnum odoratissimum, respectively – are candidates for inhibiting E protein [70].
3. HYPERINFLAMMATION, CYTOKINE STORM SYNDROME (CSS), AND SEPSIS-LIKE SYNDROME IN SARS-CoV-2 INFECTED CELLS
SARS-CoV and SARS-CoV-2 share a common transmission vector via respirable droplet dispersion and by employing the S proteins, which have a high affinity for surface receptors, such as ACE2 present in upper respiratory tract epithelial cells. The infection progresses to infect ACE2-expressing type II alveolar epithelial cells in the lower respiratory tract, advancing the disease [71]. Once the lower respiratory tracts begin to exhibit infection, there is typically the presence of pneumonitis, which can progress to create a significant disruption of the epithelial-endothelial barrier and cause unregulated and disruptive cell death through uncontrolled cellular inflammation. Also, binding of S-protein reduces the ACE2 levels, which leads to the dysfunction in renin-angiotensin system, rsulting in vascular permeability, inflammation, and the release of DAMPs to the local cellular region [72-77]. ACE2 has been shown to be present in many extrapulmonary tissues and organs, including the cardiovascular, intestinal, renal, and neuronal systems [74, 75]. The upregulated release of cytokines within the infected tissues and neighboring regions activate innate immune pathways through the release of cytokines which interact with cellular transmembrane receptors to increase activity in the NF-κB pathway, culminating in a positive feedback system in which extracellular cytokines are continually released, accelerating the local inflammation response in adjacent cell populations and resulting in what is known as CSS. CSS has been linked to the most severe forms of COVID-19 and is the cause of an extreme reaction to the viral infection, resulting in an uncontrolled immune response in which significant multi-organ damage is affected by the host immune system, causally linked to cytokine-induced inflammation, and increasing concentration of intracellular DAMPs released into the extracellular space. Given the concentration of SARS-CoV-2 in ACE2 expressing cells, and a primary infection vector through respiratory transmission, resulting pathology in more severe cases of COVID-19 are closely tied to progressive respiratory damage, and in severe cases, respiratory distress or failure know as ARDS which are a result of uncontrolled cell death from inflammatory, dysregulated apoptosis, and subsequent sepsis-like syndrome that can lead to multi-organ damage and failure [6].
CSS is primarily due to a pathogenic infection that, when dysregulated, can cause an immune system overreaction leading to detrimental systemic inflammation. Cytokine storms are the cause of some of the most severe symptoms of SARS-CoV-2 infections and can lead to fatal organ damage and mortality [ 6, 27, 30-32]. At the molecular and cellular level, the CSS begins as the virus is gaining entry to a cell and a number of Pathogen Recognition Receptors (PRRs) initiate a cascade of anti-viral immune response. Specific PRRs, such as TLRs, which can be activated by a SARS-CoV protein and trigger a signal transduction pathway that may result in activation of NF-kB, a transcription factor leading to cytokine production and cell survival, MAPK which can activate varied cell functions, including cytokine production and apoptosis, or TNFs which can activate immune cells and trigger cytokine production and cell death in neighboring cells. The viral factors may also trigger transmembrane C-type lectin-like receptors, another type of PRRs that can activate macrophages, NF-kB, MAPK and inflammasomes, multimeric protein machines that can activate IL-1β and IL-18 precipitate in pyroptosis [2, 22]. Once the virions are released inside the cell, RIG-I-like receptors (RLRs), another intracellular PRRs, may bind the viral RNA and trigger MAVS (mitochondrial antiviral signaling) cascade, activating caspase 8 and caspase 1, triggering inflammasome formation and releasing more IFNs, resulting in the killing of the infected cells and aiding in more cytokine release [2, 23]. Viral RNA can also engage another class of intracellular PRRs, NLRs, which trigger inflammasome formation, apoptosis and/or higher NF-kB-induced responses. All these cytokines, chemokines and IFN production activate local innate immune cells and recruit more host immune cells, such as lymphocytes, monocytes, and granulocytes. Neutrophils are the granulocytes that are the first to reach the site of infection to help thwart the virus or viral-infected host cell. They phagocytose viral particles or signal induction of apoptosis in infected cells. Neutrophils also release Neutrophil Extracellular Traps (NETs) and reactive oxygen species to inactivate the virus. Both are potent antimicrobial mechanisms but can also inflict local tissue damage, triggering further inflammation [5]. Macrophages and Dendritic Cells (DCs) take up and present the viral antigens to T cells [18, 78]. Other viruses have been shown to inhibit DC maturation, which prevents T cell activation and slows viral clearance [2]. It is not yet clear whether SARS-CoV-2 also has DC inhibiting proteins.
Once they bind an antigen, CD4+T lymphocytes differentiate to T helper cells which then proliferate, activate B cells with IL-4 and CD8+ T cells with IL-2 and express GM-CSF to help recruit more macrophages neutrophils, and eosinophils. In response to IL-2, CD8+ T cells are activated in killer T cells, which can trigger apoptosis in infected cells. Of note, Zhou et al found patients with COVID-19 had fewer numbers of T cells than healthy individuals as well as general lymphopenia, but those cells had higher expression of inflammatory cytokines as well as markers for cell exhaustion [31]. Fever condition enhances immune cell function, causing more efficient cytokine release [18]. This increasing cytokine environment induces CD14+ CD16+ monocytes, which express IL-6 and this IL-6 brings more leukocytes and more cytokines in an escalating loop. Specifically, in COVID-19 cases, CD14+ CD16+ monocytes have been found to express both GM-CSF and IL-6, and increased numbers of these co-expressing monocytes were associated with increased disease severity. Once in the lungs, these monocytes can differentiate into macrophages or DCs, leading to hyper inflammation [31]. These varied immune reactions occur in the context of the cellular milieu, which already existed before viral infection, and an individual’s pre-existing condition or genetic predisposition can tip the immune signaling scale away from resolution and toward the cytokine storm with its potentially debilitating consequences.
As cytokine concentration increases, as well as the release of DAMPs from ruptured cells as a result of the innate immune response to viral infection, immune cells become activated within the body and translocate to the site of infection to begin fighting off the pathogenic cause which has triggered the cellular distress call. As the SARS infection progresses, dysregulation of the immune system results in changing the immune cell responses, as well as decreasing specific cell types (e.g., CD4+ and CD8+ T Cells). Additionally, IL-10, IL-2 and TNF cytokines showed an increase in relative concentration as a function of COVID-19 disease severity progressed, reaching the highest peak during the most severe phase of the illness [30]. This disruption to immune function and depletion of functional immune cells has been suggested to have potential causative links to the disease pathology and lethality of SARS-CoV-2 through the development of viral sepsis [2].
As mentioned earlier, the immune system’s attack against an invading pathogen is the recruitment and deployment of neutrophils at the site of infection. A mechanism of attack by neutrophils is the creation of NETs. Although an effective strategy against pathogenic infection, NETs further contribute to inflammatory cell death and the progression of ARDS in patients experiencing COVID-19 through the secondary effects of further increasing the CSS, damaging tissue in increasing inflammatory apoptotic events and increasing the incidence of venous and arterial thrombosis in locales of NET formation [79], further worsening ARDS [80]. On the other hand, little is known about why lymphopenia is associated with COVID-19 as well as other CoV diseases. Aviv hypothesized that decreased hematopoiesis may result from shortened telomeres and rapid division of derived immune cells, which can lead to mitotic exhaustion [81]. Abundant myeloid-derived suppressor cells were found in patients with COVID-19, higher numbers correlating with increasing disease severity. These MDSCs were found to block T cell proliferation, but more work remains to resolve the role of these different cell types [82].
4. SEVERE COVID-19 PATIENTS EXHIBIT PATHOLOGIES IN MULTIPLE TISSUES AND ORGANS
While the respiratory system remains the primary target of SARS-CoV-2, recent evidence raise questions about the effects of infection and inflammation on other organs. Epidemiological data list cardiac injury as the most common extrapulmonary organ damaged by COVID-19, predominantly in patients with pre-existing cardiovascular conditions. Cardiomyocytes are known to express ACE2 receptors, and to some extent, TMPRSS2, and cardiovascular inflammatory markers like troponin I and D-dimer are elevated in COVID-19 patients [ 83 ] . It has been shown that COVID-19 patients are especially prone to thrombosis, with 25-30% higher occurrence of VTE and extensive micro-thrombi discovered upon autopsy. In addition, there are case reports of patients recovered from COVID-19 and released from the hospital being later treated for cardiac arrest and stroke. Thus, there is a need to understand if these findings are the result of cardiomyocytes being directly infected by SARS-CoV-2 and the molecular basis of cardiomyocyte cell death [ 84-86]. Using compiled data from the single-cell RNA-sequencing (sc-RNA-seq) studies of ACE2 and TMPRSS2 expression in various human tissues, it has been shown that the lung, large intestine and fallopian tubes are the most susceptible while the kidney, small intestine and testis are less susceptible; and the esophagus, gall bladder, brain and heart are potentially susceptible [83]. However, in a review of clinical data, it was found that heart, kidney, liver and digestive injury and tissue necrosis are common with severe COVID-19 and associated with higher mortality [86]. Evidence of viral invasion has been found in these tissues as well as nervous tissue, which may explain some of the more surprising symptoms of olfactory dysfunction [86]. Most of these findings are based on the lab work commonly performed in clinical settings to screen for acute organ dysfunction. Screening for other tissue injuries based on the data could reveal a wider effect of COVID-19. It is still unclear whether these extrapulmonary tissues are infected with SARS-CoV-2 directly or are damaged by secondary factors like cytokine storm. Interestingly, the early autopsy found scattered individual cardiomyocyte necrosis with very little lymphocyte infiltration [83, 87].
While less common, various forms of cutaneous injury have been reported since nearly the beginning of the pandemic. European studies have found a variety of manifestations in the skin, with urticaria being the first reported and erythema being most common. Others include livedo reticularis, petechiae and pruritus. In a few cases, skin injury was the first symptom noticed by the patient, but most cases of skin injury occurred later in disease progression and they found no correlation with disease severity [88]. ACE2 receptors are common in the epidermis and are also expressed in the endothelium [89]. It is yet unclear if reported skin injuries are due to the viral invasion of the skin, viral invasion of the endothelium present in the skin, or again secondary to the cytokine storm.
CONCLUSION AND FUTURE PERSPECTIVES
The plethora of results, clinical investigations, along with the detailed information and disease management directions, have been made available regarding SARS-CoV-2/COVID-19, however, the mechanisms that SARS-CoV-2 employed to hijack and manipulate host cell death pathway are still elusive. As researchers, we demand the decisions on prevention, diagnosis, and therapies to be accurate and based on the facts that are supported by reproducible data. Together, we ought to make sure that the present circumstances and progression are being accurately described and that the safety, effectiveness, and efficacy of vaccines, therapeutics, other biological products, and medical devices used for COVID-19 disease management are carefully evaluated and appraised.
LIST OF ABBREVIATIONS
| AP | = Accessory Protein |
| COVID-19 | = Coronavirus disease 2019 |
| MERS-CoV | = Middle East Respiratory Syndrome Coronavirus |
| Nsp | = Nonstructural Proteins |
| SARS-CoV | = Severe Acute Respiratory Syndrome Coronavirus |
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
This project was supported, in part, by NIH grant AI144374 (to HSC).
CONFLICT OF INTEREST
Dr. Chien-An A. Hu is the Editorial Board Member of The Open COVID Journal.
ACKNOWLEDGEMENTS
Declared none.