All published articles of this journal are available on ScienceDirect.

Identification of Naturally Occurring Antiviral Molecules for SARS-CoV-2 Mitigation
Authors Info & Affiliations
Abstract
Aim:
This study aimed to virtually screen the naturally occurring antiviral molecules for SARS-CoV-2 mitigation based on multiple molecular targets using docking and molecular dynamics simulations.
Background:
The coronavirus catastrophe (COVID-19) caused by a novel strain of coronavirus (SARS-CoV-2) has turned the world upside down at an unprecedented level and has been declared a pandemic by the World Health Organization. It has resulted in a huge number of infections as well as fatalities due to severe lower respiratory tract sickness.
Objective:
The objective of this study was the identification of inhibitors against the crucial molecular targets linked with viral infection caused by SARS-CoV-2.
Materials and Methods:
In silico screening of twenty naturally occurring antiviral molecules was performed using the Autodock docking tool. Further, molecular dynamics (MD) simulations were performed on the most stable docked complex between cysteine-like protease or 3CL protease (3CLpro) and the best-identified inhibitor (bilobetin).
Results:
3CLpros is one of the very important molecular targets as it is involved in the replication process of the virus. In the present study, we have initially investigated the inhibitory potential of naturally occurring antiviral molecules against the activity of main viral protease (3CLpro) to put a halt to viral replication. The investigation had been carried out through docking of the molecules with 3CLpro. Based on the results, the three most potential molecules (bilobetin, ginkgetin and sciadopitysin) have been screened. Further, these molecules were subjected to checking their activity on other molecular targets like papain-like protease (PLpro), spike protein S1, RNA dependent RNA polymerase (RdRp), and Angiotensin-Converting Enzyme 2 (ACE2) receptor. In addition to 3CLpro inhibition, ginkgetin was also predicted as an inhibitor of PLpro. However, none of these three compounds was found to be effective on the rest of the molecular targets. Molecular Dynamics (MD) simulations of the most stable docked complex between 3CLpro and its best inhibitor (bilobetin) confirmed notable conformational stability of the docked complex under a dynamic state.
Conclusion:
Bilobetin alone or a combination of bilobetin and ginkgetin may be used to impede viral replication. These observations are solely based on the results from blind docking with protein molecules and need to be further corroborated with experimental results.
1. INTRODUCTION
Novel coronavirus (SARS-CoV-2) was probably surfaced in a seafood market in Wuhan city of China at the end of 2019. This virus had engulfed almost the entire globe in 2020, and it still continues to do so. On February 11, 2020, it was termed COVID-19 and recognized as a cause of viral pneumonia, which had made a huge population sick. It has been declared a global health emergency and a pandemic by World Health Organization (WHO) on January 30, 2020. Since its emergence, around ~2.8 million people have died, and ~127 million have been reported being infected. Coronavirus belongs to coronviridae family (nidovirales order) and can cause respiratory illness ranging from the common cold to severe acute respiratory syndrome (SARS-CoV) with symptoms like fever, cough, and shortness of breath [1, 2]. Different coronaviruses were earlier detected in different animals like pigs, camel, bat, etc., and they may be transmitted from those animals to humans. This phenomenon is commonly termed a spillover event. Out of seven known coronaviruses, four have the ability to cause mild to moderate respiratory tract disease, and the rest of them can cause severe and lethal illnesses [3]. HCoV-OC43, HCoV-OC63, HCoV-OC229E, and HKU1 fall in the category of alpha-coronavirus, which can cause modest respiratory illnesses. The beta-coronavirus such as SARS-CoV and MERS-CoV have the potency to cause severe and fatal respiratory lower tract infection [4]. The novel coronavirus associated with COVID-19 is also a beta-coronavirus and has similarities with SARS-CoV. For that reason, it has been termed as SARS-CoV-2. In November 2002, SARS-CoV was first detected in Asia, and its further spread occurred in twenty-six countries. In September 2012, another respiratory syndrome, i.e., Middle East Respiratory Syndrome (MERS-CoV) caused by coronavirus had been reported. The coronaviruses have a large genome sequence of 30 kb in length with 5′ cap and 3′ poly-A tail [5]. The spherical virion SARS-CoV-2 has a diameter of about 60-140 nm and is constituted with peplomers of crown shape [6]. The structure mainly consists of membrane proteins, nucleoplasmid (enclosed RNA), lipid membrane, spike protein, and envelope proteins [7, 8]. The spike glycoproteins on the viral capsid play a crucial role during the internalization of the virus into the host cells. These proteins bind with the angiotensin-converting enzyme 2 (ACE2) receptor present on the surface of host cells and assist the virus in injecting RNA into cells [9]. Upon viral infection, the RNA is processed to synthesize two polyproteins (pp1a/pp1ab) [10]. The transcription process occurs through the formation of the Replication-Transcription Complex (RCT). In a typical RNA genome, there are a minimum of six Open Reading Frames (ORFs) that function as templates to produce subgenomic mRNAs. The frameshift mutation between ORF1a and ORF1b [11] encodes both pp1a/pp1ab polyproteins. The polyprotein pp1ab contains more than 7,000 residues and also possesses putative RNA-dependent RNA polymerase (RdRp) and RNA helicase activities [10, 12].
Further, these polyproteins are cleaved to form functional proteins by two proteases encoded in the virus, namely 3-Chymotrypsin-Like protease (3CLpro) or main protease and Papain-Like protease (PLpro) [13]. The key enzyme 3CLpro is the prime protease responsible for the cleavage of polypeptides into vital functional proteins required for the replication of the virus. PLpro assists the proteolytic process and also removes ubiquitin to protect viruses from immune responses [14]. Therefore, the prime protease 3CLpro, which basically generates the functional proteins required for viral replication, has attracted the attention of many researchers as a potential drug target against SARS-CoV-2 [15-20].
To identify potential drug candidates against SARS-CoV-2, we have adopted computational tools to screen out some inhibitor molecules against 3CLpro. Several reports on the identification of different potent therapeutics against SARS-CoV-2 using computational tools were published in the recent past [15-24]. In the present work, we have searched for the inhibitors in nature as several antiviral compounds were found to be present in different medicinal plants [25]. Moreover, plant-derived naturally occurring compounds play a significant role in the discovery of many effective drugs in the past, and they were approved further [26, 27].
2. MATERIALS AND METHODS
2.1. Preparation of the Structures of Small Molecules and Proteins for Docking
Three-dimensional structures (as .mol file) of twenty naturally occurring compounds and control drugs were collected from ChemSpider (www.chemspider.com). Their natural source, structures, and previously reported antiviral activities are given in Supplementary materials in Table S1. Geometry and energy optimization of their structures were performed through quantum mechanical calculations using parametric method 3 (PM3) in ArgusLab 4.0 (http:// www.arguslab.com). The crystal structure of the SARS-CoV-2 related proteins, namely 3CLpro (PDB ID: 6M0K), PLpro (PDB ID: 6W9C), RdRp (in a complex with SARS-CoV-2 NSP7 and NSP 8, PDB ID: 6M71), spike protein S1 (in complex with human antibody, PDB ID: 6W41), ACE2 (in a complex with spike glycoprotein, PDB ID: 6LZG) were downloaded from Protein Data Bank (PDB). To refine these protein structures, bound ligands or proteins and the crystallographic water molecules were removed from the structures.
2.2. Molecular Docking
Protein-ligand dockings were performed by using Autodock 4.2. Before docking, hydrogens were added, torsion angles were confirmed, and Kollman charges were added to the protein structure. The grid boxes for the blind docking were made in such a way that the whole protein in each case was enclosed within that box. Further, the Lamarckian Genetic Algorithm (LA) protocol was applied in the docking. The lowest energy docked conformation of a ligand obtained from each docking was saved as a .pdb file. That conformation of ligand was merged with the corresponding protein structure, and then that was used for the analysis of protein-ligand interactions. Interacting residues of the proteins along with the types of interactions were identified using Protein-Ligand Interaction Profiler (https://projects.biotec.tu -dresden.de/plip-web/plip). Molecular visualization and rendering of the structures were done in Pymol.
2.3. Determination of logP Value
The logP values of the compounds were predicted using the SWISSADME (www.swissadme.ch/index.php) server.
2.4. Molecular Dynamics (MD) Simulation
3CLpro-bilobetin complex (as bilobetin appeared with the highest docking score with 3CLpro) was selected to probe binding stability. For this purpose, MD simulation of the complex was performed over a period of 150 ns. Here, we had used SPC water model using the GROMOS54A7 force field executed in Gromacs 5.1.2 [28]. A prior simulation setup containing protein-ligand complex was developed and pre-equilibrated under biological environment. 0.15 M salt concentration and 4 Na+ were used for neutralizing the simulation setup in the cubic simulation box. For energy minimization of the protein-ligand complex in the arranged system, the steepest descent method for 50,000 steps was employed. Using the SHAKE algorithm, the equilibration runs for the system were performed under NPT and NVT conditions for 1 ns each. To maintain the average temperature and pressure at 300K and 1 bar, we used the V-rescale thermostat (a modified algorithm of Berendsen thermostat) and Parrinello-Rahman barostat methods, respectively.
2.5. Analysis of MD Simulation
The Root Mean Square Deviation (RMSD), Root Mean Square Fluctuation (RMSF), and the Radius of gyration (Rg) for C-α atoms were calculated using gmx, rms, rmsf, and gyrate commands, respectively. Global conformational analysis as derived from the Principal Components (PC) of the system was used to estimate almost the whole dynamics of the system. It was measured using covar and anaeig commands in Gromacs. Solvation (polar and non-polar) free energy and molecular mechanics (mm) potential energy (electrostatic and Van der Waals energy) were calculated using g_mmpbsa tool [29, 30].
3. RESULTS AND DISCUSSION
To restrict the spread of COVID-19 infections, inhibition of 3CLpro seems to be a potential way to discontinue the process of viral replication. Many recent articles have focused their target on 3CLpro to fight SARS-CoV-2, as mentioned earlier in this article. Considering the importance of natural compounds, we have selected twenty naturally occurring antiviral molecules, and they were docked with 3CLpro using Autodock for screening their potential. In addition to that, we had also docked some control drugs (remdesivir, lopinavir, ritonavir, and ribavirin), which are under some preclinical and clinical trials against SARS-CoV-2. Based on the clinical and preclinical reports as obtained from in vitro animal models as well as clinical studies of the drugs such as remdesivir, hydroxychloroquine, lopinavir, and ritonavir, their emergency use was approved in 2020 for the treatment of COVID-19 [31].
Further clinical studies reported mixed outcomes on the effectiveness of these drugs [ 31 ]. Their efficiency also depends on the clinical protocol frameworks. In the present study, we have chosen those drug molecules as control drugs to have a comparative idea about the effectiveness of our selected test molecules. The free energy of binding all these molecules with 3CLpro as estimated by Autodock is given in Table 1.
| S. No. | Compound | Estimated Free Energy of Binding with 3CLpro (kcal/mol) |
Estimated Inhibition Constant for 3CLpro (μM) |
logPO/W (consensus) |
|---|---|---|---|---|
| 1. | Bilobetin | -10.83 | 0.011 | 3.96 |
| 2. | Ginkgetin | -10.19 | 0.033 | 4.34 |
| 3. | Sciadopitysin | -9.20 | 0.180 | 4.76 |
| 4. | Narasin | -5.18 | 159.86 | 5.20 |
| 5. | Resveratrol | -5.83 | 53.61 | 2.48 |
| 6. | Esculetin | -5.90 | 47.30 | 1.12 |
| 7. | Esculin | -5.30 | 130.02 | -0.56 |
| 8. | Matrine | -7.41 | 3.71 | 1.80 |
| 9. | Scutellarin | -5.07 | 193.03 | -0.22 |
| 10. | Delphinidin | -6.13 | 31.88 | 0.13 |
| 11. | Cyanidin | -6.19 | 28.87 | 0.56 |
| 12. | Pelargodin | -6.73 | 11.60 | 2.70 |
| 13. | Harmine | -6.02 | 38.79 | 2.78 |
| 14. | Harmane | -5.47 | 97.57 | 2.70 |
| 15. | Harmol | -6.35 | 22.07 | 2.16 |
| 16. | Avarol | -7.35 | 4.08 | 4.75 |
| 17. | Avarone | -7.93 | 1.54 | 4.24 |
| 18. | Polyandrocarpidine B | -5.49 | 94.61 | 2.76 |
| 19. | Polyandrocarpidine D | -5.83 | 53.60 | 2.33 |
| 20. | Halitunal | -6.30 | 23.94 | 4.54 |
| 21. | Remdesivir | -4.35 | 644.64 | 1.53 |
| 22. | Ritonavir | -3.26 | 4060 | 5.03 |
| 23. | Lopinavir | -4.14 | 919.24 | 4.53 |
| 24. | Ribavirin | -4.38 | 373.99 | -2.05 |
We also calculated the logP value of these compounds to check their drug likeliness. Basically, when a solute is distributed between two solvents, the ratio of the concentration of that solute in those solvents is termed partition coefficient (P). If a compound is partitioned between water and a nonpolar solvent, the logP value is considered as a measure of lipophilicity or hydrophobicity of that compound. In general, the concentrations of the solute in the nonpolar and polar phase are placed in the numerator and denominator, respectively, to determine the value of P. The logP value or lipophilicity is a crucial parameter to understand the cell penetration behavior of a molecule through cell membranes. If logP value is more than 5, it suggests reduced absorption and less permeability due to greater molecular hydrophobicity [32]. The logP values of all these compounds are enlisted in Table 1. Except for narasin, all of our selected molecules have an estimated logP value of less than 5.
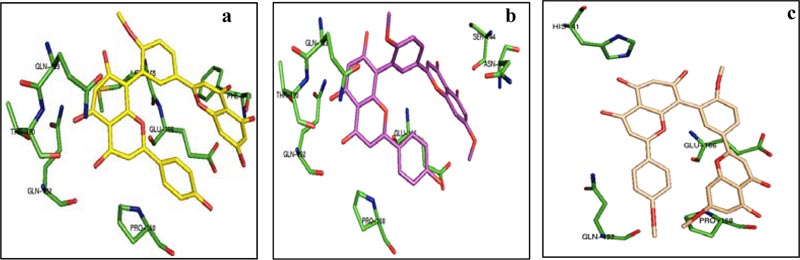
From the above table, it was found that the estimated ΔG is very high for bilobetin (-10.83 kcal/mol), ginkgetin (-10.19 kcal/mol), and sciadopitysin (-9.20 kcal/mol). Other molecules have the binding energy in the range of -5.07 to -7.93 kcal/mol. The estimated ΔG values for the control drugs remdesevir, ritonavir, lopinavir, and ribavirin are -4.35, -3.26, -4.14, and -4.38 kcal/mol, respectively. Based on these values, three molecules from our series, namely bilobetin, ginkgetin, and sciadopitysin were found to be promising inhibitors of 3CLpro. We have further extended our study to trace the nature of interactions between 3CLpro and these three molecules. The lowest energy docked conformation of these compounds with 3CLpro is shown in Fig. (1). The residues of 3CLpro, which strongly interact with bilobetin, ginkgetin, and sciadopitysin are also marked in Fig. (1). Different non-covalent forces such as hydrogen-bonding, hydrophobic, Van der Waals, π-stacking interactions, etc., were found to be involved in protein-ligand interactions. The substrate-binding site of 3CLpro is constituted by the residues Thr 25, Thr 26, His 41, Met 49, Gly 143, Cys 145, Glu 166, Pro 168, etc. A recent report has revealed the role of two catalytic residues, namely His 41 and Cys 145, along with some other residues like Gly 143, Cys 145, His 163, His 164, Glu 166, Pro 168, and Gln 189 for effective design of suitable inhibitors with 3CLpro [33]. The importance of these residues in the design of antiviral compounds as inhibitors of 3CLpro was also supported in another recent publication [34]. Bilobetin (Fig. 1A) was found to form seven possible H-bonds with the residues Phe 140, Glu 166, Gln 189, Thr 190, and Gln 192. It also has three hydrophobic interactions with Met 165, Glu 166, and Pro 168. Ginkgetin (Fig. 1B) interacts with the residues Asn 142, Ser 144, Glu 166, Gln 189, Thr 190, and Gln 192 through nine H-bonds and with Pro 168 through hydrophobic interaction. Sciadopitysin (Fig. 1C) is involved in three H-bonding with His 41 and Gln 166 and five hydrophobic interactions with Glu 166, Pro 168, and Gln 192. The catalytic residue His 41 is 4.13 and 2.92Å away from bilobetin and sciadopitysin molecules, respectively. In contrast, the distance between ginkgetin and the other catalytic residue Cys 145 is 2.92Å. In addition to these interactions, the inhibitor molecules may also have π-cation, π-stacking, and Van der Waals interactions.
Therefore, blind docking of the naturally occurring antiviral molecules with 3CLpro suggests that the above-mentioned three compounds possess excellent inhibitory potential (with nanomolar inhibition constant) towards this protease enzyme. This is because of their strong binding at the catalytic site of the enzyme, which is crucial for viral replication. In the case of four control drugs, 3CLpro was not predicted as a suitable molecular target in terms of the binding energy values mentioned in Table 1. The residues of 3CLpro interacting with these control drugs are also mentioned in Table 2. Among them, only ribavirin was found to bind at the catalytic site of 3CLpro.
| Compound | Residues of 3CLpro Interacting with the Molecule |
|---|---|
| Remdesivir |
Hydrophobic interactions: Arg 4, Lys 5, Met 6, Ala 7, Val 125, Tyr 126 Hydrogen bonding: Lys 5, Ala 7, Gly 127, Arg 298 |
| Ritonavir |
Hydrophobic interactions: Lys 5, Ala 7, Val 125, Tyr 126, Gln 127, Glu 288, Phe 291 Hydrogen bonding: Lys 5 |
| Lopinavir |
Hydrophobic interactions: Tyr 239, Met 276, Ala 285 Hydrogen bonding: Leu 271, Gly 278, Ala 285 |
| Ribavirin | Hydrogen bonding: His 164, Gln 189, Thr 190, Gln 192 |
3.1. Molecular Dynamics Simulations
MD simulations were performed to investigate the binding stability of topmost screened compound (bilobetin) with 3CLpro of SARS-CoV-2. Bilobetin, out of screened 24 compounds, had shown a significantly high binding energy with 3CLpro. The extensive MD simulation up to 150 ns had been analyzed in terms of mean deviation, fluctuation, and radius of gyration of the system. As observed in the trajectory, the 3CLpro complex with bilobetin had shown an increasing trend (0.2-0.3 nm) in RMSD values up to 60 ns while it has been stabilized after 100 ns with slight fluctuations in the middle (Fig. 2A). The average RMSD of the complex was calculated as ~0.28 nm for 150 ns simulation time which is in a favorable range. The mean fluctuation (measured as RMSF) was observed to be in good correlation with the RMSD of the complex.
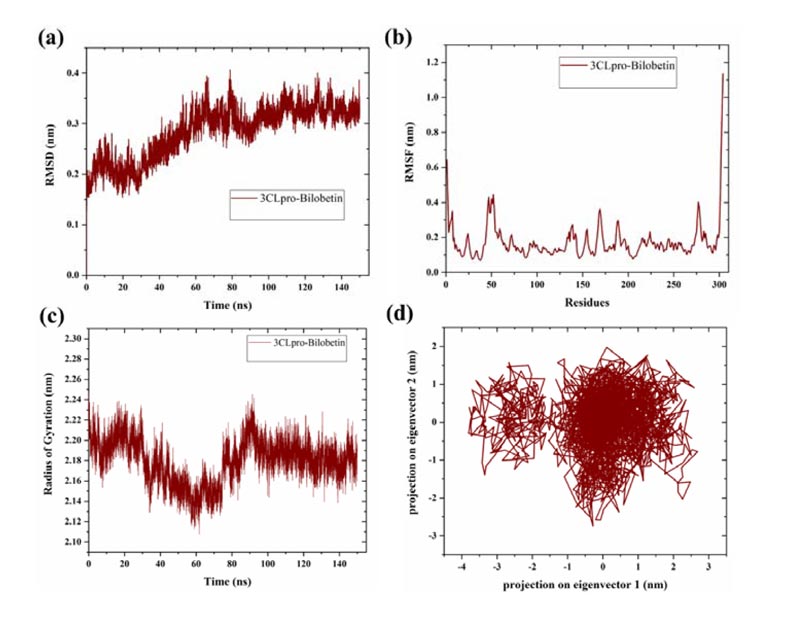
3CLpro-bilobetin complex revealed slightly higher fluctuation upto 0.4 nm near residues 46-53 and the rest of the protease did not show such fluctuations (Fig. 2B). An intense peak was also detected at C-terminal, which may be due to the presence of disordered residues at the terminal [35]. The radius of gyration (a parameter to measure biomolecule compactness) was also determined to study the effect of the binding of bilobetin on the structure of 3CLpro. Like RMSD and RMSF, bilobetin-3CLpro complex had shown fluctuations till nearly half time of the simulation and then attained stability after 85 ns with a lesser radius. The average radius of gyration of bilobetin bound complex was calculated to be approximately 2.21 nm for 150 ns long MD simulations (Fig. 2C). Lesser value of the radius of gyration suggests more compact structure of the complex. This also indicates that bilobetin could bind to 3CLpro with significant binding efficacy. As a conclusion, these three parameters had suggested quite stable binding of bilobetin with 3CLpro but that needs to be validated through experimental studies.
3.2. Principal Component Analysis (PCA)
Principal components or principal motions are the main and responsible components of a trajectory to determine the overall variations that occurred during the simulation time. The conformational alterations can be identified through the PCA for the C-α atoms of the protein. To investigate the overall change in the structural conformations, we have analyzed the simulation trajectory through these principal motions. Using the normalized data from the trajectory, a co-variance matrix of eigenvalues and eigenvectors was created from first two components that define the overall motion in the system [24061923, 32457393, 31443266]. PCA generates eigenvectors which represent the reduced collective atomic motions in a protein structure [36]. The last 25 ns of the simulation trajectory was analyzed and it was found that the bilobetin bound 3CLpro complex had occupied a less conformational space, as projected with two principal components (Fig. 2D). A cluster is formed by eigenvectors with a distance range -1 to 2 nm and a scattered region is shown at -3.5 to -2 nm. Overall, significantly effective binding of bilobetin indicates that it could act as a potential druggable molecule against SARS-CoV-2.
3.3. Binding Energy Estimation
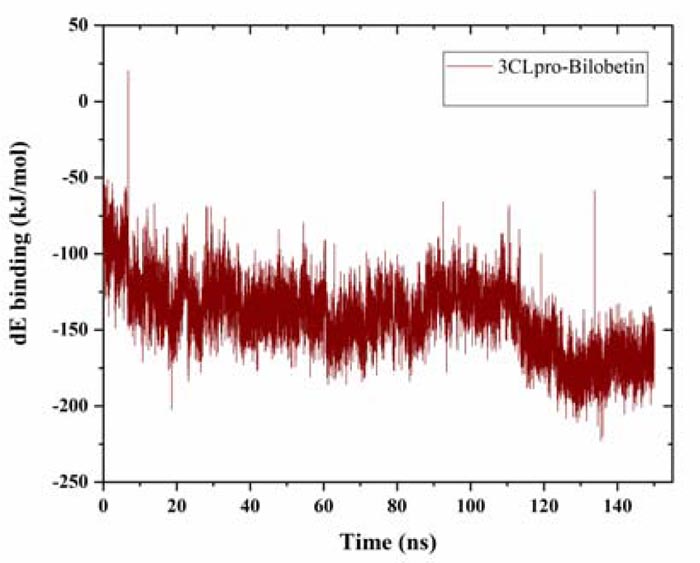
The binding energy was estimated for the whole trajectory. Using g_mmpbsa tool, we had calculated the solvation (polar and non-polar) free energy and molecular mechanics (mm) potential energy (electrostatic and van der waals energy) [29, 30]. The average binding energy was estimated to be -141.38 ± -23.91 kJ/mol, which is quite acceptable (Fig. 3). The solvent accessible surface area (SASA) energy was found to be -21.15 ± -1.58 kJ/mol. The estimated binding energy value also supports the observations from MD simulation and strongly suggests further validation of the protein-ligand binding through in vitro experiments.
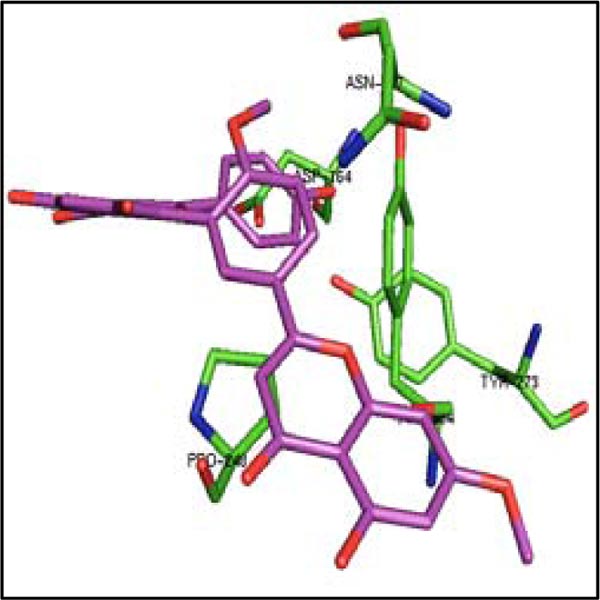
Considering admirable inhibitory capability of these three molecules on 3CLpro, their binding was also studied with another protease PLpro of SARS-CoV-2. The binding of four control drugs (remdesivir, liponavir, ritonavir, and ribavirin) with PLpro was also checked. In Table 3, it has been found that the estimated ΔG is very high for bilobetin (-10.83 kcal/mol), ginkgetin (-10.19 kcal/mol), and sciadopitysin (-9.20 kcal/mol). The catalytic residues Cys 111 and His 272 (residue numbering according to the pdb file) of the active site of PLpro are present in S1 pocket. The substrate binding site is most probably located in the S3/S4 pockets, which are much more spacious than the S1/S2 pockets situated very close to the catalytic residues [37]. The residues from Asp 164 to Glu 167, Met 208, Cys 217, Ala 246 to Pro 248, Tyr 264, Gly 266 to Gln 269, Gly 271, Tyr 273, Thr 301 and Asp 302 are present in the substrate binding region of PLpro [37]. Considering the residues of PLpro interacting with these three molecules (Table 4), it was noticed that only ginkgetin can bind in the S3/S4 pockets (Fig. 4). This molecule interacts closely with the residues of that pocket as mentioned above. Therefore, ginkgetin is expected to inhibit the proteolytic activity of PLpro as its binding in that region can inhibit the enzymatic activity of PLpro.
| S. No. | Compound | Docking with PLpro | Docking with Spike protein S1 | Docking with ACE2 | Docking with RdRp | ||||
|---|---|---|---|---|---|---|---|---|---|
| Binding energy (kcal/mol) |
Inhibition constant (μM) |
Binding energy (kcal/mol) |
Inhibition constant (μM) |
Binding energy (kcal/mol) |
Inhibition constant (μM) |
Binding energy (kcal/mol) |
Inhibition constant (μM) |
||
| 1. | Bilobetin | -9.63 | 0.088 | -11.13 | 0.007 | -9.91 | 0.054 | -9.49 | 0.11 |
| 2. | Ginkgetin | -6.81 | 10.11 | -11.23 | 0.006 | -7.96 | 1.46 | -9.78 | 0.067 |
| 3. | Sciadopitysin | -9.44 | 0.121 | -11.31 | 0.005 | -8.90 | 0.298 | -9.26 | 0.162 |
| 4. | Remdesivir | -2.73 | 9910 | NP | NP | NP | NP | -3.52 | 2630 |
| 5. | Ritonavir | -3.29 | 3860 | NP | NP | NP | NP | NP | NP |
| 6. | Lopinavir | -4.51 | 497.85 | NP | NP | NP | NP | NP | NP |
| 7. | Ribavirin | -3.95 | 1280 | -5.03 | 205.19 | NP | NP | -3.32 | 3670 |
| 8. | Hydroxychloroquine | NP | NP | NP | NP | -5.77 | 58.63 | NP | NP |
| Compound | Residues of PLpro interacting with the molecule | Residues of Spike protein S1 interacting with the molecule | Residues of ACE2 receptor interacting with the molecule | Residues of RdRp interacting with the molecule |
|---|---|---|---|---|
| Bilobetin |
Hydrophobic interactions: Arg 65, Val 66, Ala 68, Phe 69, Thr 74, Thr 75, Phe 79, Leu 80 π-stacking: Phe 69 |
Hydrophobic interactions: Phe 342, Ala 363, Val 367, Leu 368, Phe 374 Hydrogen bonding: Cys 336, Glu 340, Asn 343, Asp 364, Tyr 365, Val 367, Leu 368 |
Hydrophobic interactions: Leu 95, Asn 210 Hydrogen bonding: Trp 203, Asp 206, Asn 210, Ala 396, Glu 398, Arg 514, Glu 564, Trp 566 |
Hydrophobic interactions: Tyr 273, Leu 329, Val 330, Arg 331, Lys 332, Val 341 Hydrogen bonding: Leu 270, Pro 328, Val 330, Arg 331, Thr 344 |
| Ginkgetin |
Hydrophobic interactions: Asp 164, Pro 248, Tyr 264 Hydrogen bonding: Asp 164, Asn 267, Tyr 273 |
Hydrophobic interactions: Pro 337, Phe 342, Ala 363, Val 367, Leu 368, Phe 374 Hydrogen bonding: Cys 336, Glu 340, Asp 364, Tyr 365, Val 367, Leu 368, Ser 371 |
Hydrophobic interactions: Asn 290, Ile 291, Leu 370, Phe 438 Hydrogen bonding: Asp 292, Asp 367, Phe 438, Gln 442 π-cation: Lys 441 |
Hydrophobic interactions: Arg 249, Val 315, Leu 460, Pro461 Hydrogen bonding: Ala 250, Leu 251, Ser 255, Thr 319, Arg 349, Phe 396, Asn 628 |
| Sciadopitysin |
Hydrophobic interactions: Tyr 213, Glu 214, Tyr 305, Lys 306 Hydrogen bonding: Lys 217, Glu 307 |
Hydrophobic interactions: Pro 337, Phe 338, Glu 340, Phe 342, Val 367, Leu 368, Phe 374 Hydrogen bonding: Cys 336, Glu 340, Asp 364, Tyr 365, Leu 368 π-stacking: Phe 342 |
Hydrophobic interactions: Ala 99, Gln 102, Glu 398, Lys 562 Hydrogen bonding: Ala 99, Gln 102, Glu 208, Asn 394 π-cation: Lys 562 |
Hydrophobic interactions: Tyr 420, Leu 437, Phe 440, Phe 843 Hydrogen bonding: Gly 413, Tyr 420, Glu 436, Leu 437, Phe 441 π-stacking: Phe 415, Phe 843 |
A major hotspot is recently identified in the spike protein S1 of SARS-CoV-2 for its binding with ACE2 receptor [38]. This binding region in the spike protein is composed of Lys 417, Asn 487, Gln 493, Gln 498 and Tyr 505. Another recent article [23] also identified the residues Tyr 453, Arg 454, Leu 455, Lys 458, Ser 459, Ser 469, Glu 471, Pro 491, Leu 492, Gln 493 and Tyr 489 as a part of binding site. The values of estimated free energy of binding with spike protein S1 are highly negative in case of these three molecules (Table 3). However, the binding site for bilobetin, ginkgetin and sciadopitysin (interacting residues enlisted in Table 4) in the spike protein is quite different than the predicted hotspot for receptor binding. In this case, these molecules probably will not be effective in preventing the binding of the spike protein with its receptor on host cells. Similarly, the binding hotspot in ACE2 receptor is composed of Lys 31, His 34, Glu 35, Glu 37, Asp 38 and Try 83 [38]. In this case also, none of the three molecules bind in that region of ACE2 to prevent its binding with spike protein S1 of SARS-CoV-2 with ACE2. In case of RdRp, two aspartic acid residues namely Asp 760 and Asp 761 (residue numbering as per pdb file) constitute the active site [22, 39]. From Table 4, it is also clear that these three molecules do not bind to the active site of RdRp.
CONCLUSION
Using docking tool, three amentoflavone (bilobetin, ginkgetin and sciadopitysin) were predicted to inhibit the main protease (3CLpro) of SARS-CoV-2, which is very important for viral replication. Molecular dynamics simulation analysis revealed that the highest scoring inhibitor bilobetin can form a complex with 3CLpro with high conformational stability. Among these molecules, ginkgetin was also identified as an inhibitor of Papain-Like protease (PLpro) of SARS-CoV-2. When these three promising molecules were docked with other molecular targets associated with SARS-CoV-2 (spike protein S1, RNA dependent RNA polymerase and Angiotensin Converting Enzyme 2 (ACE2) receptor), it was observed that they do not bind to the active sites or hotspots on those protein targets. These observations are solely based on the results from blind docking with protein molecules and they need to be further corroborated with experimental results for a fruitful conclusion.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
Not applicable.
HUMAN AND ANIMAL RIGHTS
No animals/humans were used for studies that are the basis of this research.
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of this study are available within the article.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
KSG is grateful to the Director, NIT Hamirpur for research supports. Authors are thankful to Prashant Yadav, Navdeeshwar Suman, Ayushi Aggarwal, and Khusboo Kumari (UG students from NIT Hamirpur) for their support in some docking experiments. AS is thankful to NIT Hamirpur for her fellowship.
SUPPLEMENTARY MATERIAL
Supplementary material is available on the publisher’s website along with the published article.

