All published articles of this journal are available on ScienceDirect.

A Focused Review on Emerging SARS-Cov-2 Variants
Authors Info & Affiliations
Abstract
Over the past 33 months, 10 SARS-CoV-2 mutant variants have evolved. Among them, very few were infectious, while the rest were not that rampantly infectious. As COVID-19 has been the century’s most destructive pandemic, and as the variants of concern and significance are heralding nature, there is an inherent need to bring collaborative convergence among scientists to combat future pandemics such as COVID-19. A question remains whether these emerging variants could allow us to define the advent of pathogenesis and whether or not we are prepared. In this review, we give a gist of variants that could be associated with the recombination events concerned with SAR-CoV-2 that have an impact on the immune response in the human body.
1. INTRODUCTION
SARS-CoV-2 has been one of the most discussed and extensively researched topics over the past few years. It has a genome size of about 29.9 kb and is a single-stranded positive-sense RNA virus. The origins of the unique pandemic coronavirus had their inception in December 2019 in Wuhan even as It exhibits the phenomenon of genetic drift [1, 2]. Due to the lack of a definite origin, the evolution of the origin of SARS-CoV-2 is still unclear. Whether the origin of the virus is due to the phenomenon of natural selection in an animal host before zoonotic transfer or the same phenomenon in humans after zoonotic transfer remains elusive [3-5]. SARS-CoV-2 causes severe respiratory illnesses such as lung failure and pneumonia and over the last 33 months, t even as the flagship genes have begun to be studied even as it has been unfolding the saga of COVID-19 infection [6, 7].
Viruses cause significant morbidity as well as mortality in humans because they are generally deadly, proliferate, and possess a mechanism of their own. Effective drugs that can control their proliferation are currently unavailable on the market. Three of the seven known human coronaviruses, namely SARS-CoV, MERS-CoV, and SARS-CoV-2 that have developed and evolved into their respective severe pathogenic forms are infecting and colonizing the human respiratory tract. Droplets, aerosols, and contaminated fomites are the predominant contact routes potentially responsible for viral spread [8]. Some patients were identified with mutations representing multiple viral strains, which may have formed as a result of homologous recombination due to co-infection. Major viral strains with nucleotide substitutions may exist between Wuhan-1 and RaTG13 coronavirus as the genome tends to switch between these two viruses and thus shows high instability or mutation tolerance [9]. On the other hand, SARS-CoV-2 and its ancestors, SARS-CoV and bat-SARS-like-CoV, possess a shared origin. A thorough analysis of Codon Usage Bias (CUB) in SARS-CoV-2 revealed significant CUB, natural selection, and mutation pressure dominating in the S-genes of distinct beta-coronaviruses [10].
Betacoronaviridae's infectivity and virulence fluctuate over time due to their extremely high mutation rate, frequent recombination, and interspecies transmission. SARS-CoV-2 genotypes revealed mutations in structural, non-structural, accessory proteins and untranslated regions. However, the most common type of mutations among the many available are substitutions of single nucleotides, including insertion or addition, removal or deletion, as well as frameshifts, at a slower rate. Out of all the structural proteins, the spike glycoprotein as well as the nucleocapsid phosphoprotein, show the most changes whereas proteins in the envelope and the proteins in the membrane turn out mostly to be conserved. Variations in D614G, as well as P323L, proteins like the spike, and also the enzyme RNA-dependent RNA polymerase, became dominant globally [11], suggesting a foundation to understand the recombination apart from the noise and convergent evolution with recombinations happening before its transfer to humans [12].
Humans were first infected with SARS-CoV in 2002. Many believe that the Carnivora-borne viruses are most likely to be the source of SARS-CoV. While the development of the MERS-CoV in 2012 was likewise aided by Chiroptera-hosted lineages, the causal microorganism of COVID-19, SARS-CoV-2 has been added to zoonotic transmission events from 2019. Coronavirus was studied as outgroups across eleven different species, SARS-CoV, SARS-CoV-2, and MERS-associated viruses are all connected to the Chiroptera-hosted viruses [13]. All current Coronaviridae members are the result of a series of gradual adaptive modifications that occurred through multiple previous recombination events, resulting in each having an RNA sequence with a unique mosaic pattern. The core sequence corresponding to the novel coronavirus, known as SARS-CoV-2, was acquired from a parent common to a bat coronavirus, portrayed by the strain RaTG13, which was discovered in 2012 in the Yunnan province of China [2]. The genetic similarity of Beta coronavirus found in Rhinolophus bats in China to the variant of SARS-CoV-2 was profusely elevated, but they are not likely to be the direct progenitor [14]. Whereas Alpha CoV was identified as a co-infection in one of the bat samples, close observation is needed in bats to evaluate the degree of risk of newly arising CoVs [15]. There were two major variants, one of which was present in the initial epidemic in Wuhan and the latter, the S-type which is older and emerged previously than L-type and less pervasive at early stages [16].
1.1. Sequence and Evolutionary Lineages of SARS-CoV-2 in the Realm of Immunobiology and Drug Efficacy
Over the last few months, drug and vaccine development saw failures as new strains with altered virulence and the ability to escape antiviral defense or repurposed drugs. The new strains that emerged due to rapid recombination are likely to target the immune system [17]. Antigen receptors playing an important role in the innate immune system, such as nucleic acids, carbohydrates, lipids, small molecules, and proteins that are specific, have intrinsic specificity, which has been described by antigen receptors present as part of the innate immune system. These two immune system components either react quickly (innate) with specific antigens of microbes or evolve through time (adaptive) in order to reach greater specificity as well as high affinity. Other important receptors of the immune system, like the cytokine receptors, which help in regulating immunity as well as inflammation, are also present. Antigen receptors utilize a minimal number of protein folds to fulfill their several varieties of immunological tasks with other essential participants, the antigens themselves. Surface glycoproteins which are present in enveloped viruses, such as SARS-CoV-2, allow access into host cells and also are the targets for antibody responses [18]. Divergent genetic polymorphisms created a significant challenge in developing medicines and vaccines [10].
In the SARS-CoV-2 pandemic, a very important role is executed by the adaptive immune system and influences a patient’s clinical outcomes. A delayed and weak adaptive immune response was seen in patients who had critical COVID-19 symptoms. It has been reported that the repair mechanism for DNA damage, needed for the mechanism of V(D)J recombination in adaptive immunity, was inhibited by the spike protein of SARS-CoV-2 by using an in vitro cell line [19]. It has also been found that the nucleus is where the spike protein localizes and impedes BRCA1 and 53BP1 DNA repair protein recruitment to the particular damage site. The studies revealed a possible molecular mechanism for more understanding this mechanism [19]. The SARS-CoV-2 genome acts as a messenger RNA and translates open reading frames (ORFs), viz. polyproteins of ORF1a and ORF1b regions, that get cleaved into a number of 16 non-structural proteins (nsp1–16) by proteases present in the virus, and initiates viral replication and transcription after the virus enters the host cell expressing both ACE2 and TMPRSS2 receptors. Many ORFs could be better annotated and predicted if the subgenomic sequences were translated into hypothetical proteins [20].
In Yunnan province, a novel SARS-CoV-2-related virus, provisionally known as PrC31, was found by examining the next-generation sequencing (NGS) data of intestinal samples collected in 2018. The nucleotide identities of the SARS-CoV-2 and SARS-CoV-2 ZC45 genomes were 90.7 and 92.0 percent, respectively. The genomic regions of PrC31 with the highest levels of similarity to the equivalent genomic regions of SARS-CoV-2 were orf1a and orf8, in particular. Recombination investigations reveal that PrC31 originated from yet-to-be-identified intermediate recombination strains and underwent numerous complex recombination events involving the SARS-CoV and SARS-CoV-2 sub-lineages [21]. SARS-CoV-2 is mostly found to be chimeric, as revealed by genomic analyses. They are similar to the RaTG13 sequence of CoV, but again, its RBD is similar to pangolin CoV. Viruses that are chimeric can occur through either human interference or natural recombination [22]. Civets and camels are intermediate hosts, allowing such viruses to recombine and appear as fresh pathogenic and aggressive strains [23]. The authors have also identified the SARS-CoV-2-related coronaviruses in Malayan pangolins. Viruses linked to SARS-CoV-2 that are connected with pangolins have been discovered, implying that pangolins could also be the possible hosts of new coronaviruses [24]. The virus isolated from Malayan pangolin, referred to as pangolin-CoV, has a high amino acid identity with that of SARS-CoV-2. The pangolin-CoV’s receptor-binding domain of the S protein is similar to SARS-CoV-2. 17 out of the 25 Malayan pangolins had pangolin-CoV. Pangolins that were infected had clinical symptoms, and antibodies against the pangolin-CoV reacted with SARS-CoV-2’s S protein. As noticed, pangolins could also be the intermediate hosts of SARS-CoV-2, and a series of recombination studies have been acknowledged [25-32].
In the genomes of numerous SARS-CoV-2 strains, deletions have been discovered that cannot be resolved using existing phylogenetic approaches. As a result, employing strict phylogenetic remodeling, the k-mer natural vector is supposed to investigate hosts and transmission features for SARS-CoV-2. The fact that SARS-CoV-2 clusters with bat-origin coronaviruses, strongly supports the fact that bats are the virus's natural reservoir. After creating a bat-to-human transmission route, the civet is projected as a potential alternative, and the pangolin is acknowledged as an intermediary host [33]. It is important to comprehend the origin of SARS-CoV-2 in order to prevent future zoonoses [34]. The SARS-CoV-2 pandemic and numerous other zoonotic epidemics in humans highlight the significance of researching the evolution of the entire CoV subfamily to comprehend how new strains arise and what molecular processes affect their adaptation, transmissibility, host/tissue tropism, and pathological non-homologous genetic structural restrictions that prevent the SARS-CoV-2 spike from evolving further. Point mutations, insertions/deletions, and intra-SARS-CoV-2 recombination events lead to the emergence of novel SARS-CoV-2 strains [35]. HIV-1 and SARS-CoV-2 have the same evolutionary strategies. The variability generated by recombination and mutations enables extraordinary diversity in HIV-1 and a slower rate of mutation in SARS-CoV-2. Important phenotypic consequences have been shown by recent variants of SARS-CoV-2, exhibiting spike mutations that have improved infectivity and antibody resistance. The authors have compared mutational patterns in HIV-1 and understanding the diversity of SARS-CoV-2 that leads to their evolution by natural selection [36], besides understanding the structures [37] and their variations over time [38] has been helpful in many ways for the analysis of antiviral drugs [39]. Novel variant surveillance [40] and a substantial impediment to the creation of CoV-specific vaccines and medicines [41-43]. A new strain may evolve due to rapid recombination and the ability of independent species to cross. In order to battle SARS-CoV-2, it is vital to educate the public about personal hygiene, social isolation, and the use of available treatments. In order to prevent viral spillover outbreaks, it is necessary to identify potential sources, intermediate hosts, and the virus's ability to cross species barriers [44].
1.2. Spike as a Key, Important Structural Gene: Are there any known Recombination events Occurring in COVs?
Coronaviruses have a special ability for genetic recombination and greater mutation rates, which result in adaptations that make them better suited to overcome species barriers and infect other species. High homologous RNA recombination rates, low fidelity of the viruses' RNA-dependent RNA polymerase, and adaptation of the S protein to bind host receptors, such as angiotensin-converting enzyme 2 (ACE2) in the case of SARS-CoV and SARS-CoV-2, and dipeptidyl peptidase 4 (DDP4) in the case of MERS-CoV, are all factors that contribute to the viruses' high diversity [45].
The spike gene has several variants, some of which are shared across or between strains. The alpha strain is vulnerable to immunological responses caused by previous strains, the beta, gamma, and delta strains, which are immune-resistant. Aside from random replication errors, intra-host RNA editing, chronic infections, and recombination are all factors that can cause sequence alterations to accumulate in the SARS-CoV-2 genome. Recombination has a lead role in coronavirus evolution, and recent results on SARS-CoV-2 imply that it might be especially important [46]. The latest SARS-CoV-2 variation of concern, Omicron, has drawn attention to step change antigenic occurrences, or “shifts,” rather than incremental “drift” changes in antigenicity [40]. Several spike protein and RBD mutations arose in many variations, with over 30 alterations in the new Omicron (B.1.1.529) clade, 15 of which were focused on the RBD [47]. The molecular and evolutionary mechanisms that control the cell tropism and interspecies transmission of coronaviruses have started to reveal after SARS-CoV, MERS-CoV, SADS-CoV and SARS-CoV-2. Carbohydrates and protein receptors, to which the spike protein binds, induce membrane fusion, and these interactions are so complex that even after many improvements in the field, there are many unanswered concerns regarding tropism modification and cross-species transmission. The evolution of spike has largely been concentrated because it is highly susceptible to mutations and recombinations. Millet JK et al . (2021) brings light to the molecular and evolutionary insights of coronavirus receptor usage and host range expansion. Multiple recombination events have been harbored by SARS-CoV-2 genomes. S proteins have the ability to infect humans and mammalian cells as they show purifying selection and ancestral recombination events. Future recombination opportunities increase as the SARS-CoV-2 population grows and spreads globally [48]. Scientists have found many independent origins for recombinant SARS-CoV-2 viruses carrying single-nucleotide polymorphisms and B.1.1.7 variant characteristic deletions. Genomes of recombinant strains share genetic variation that is contiguous with non-B.1.1.7 viruses present in the same place as the recombinants. Recombination breakpoint location suggests that community-transmitted recombinant viruses obtained their spike area from a B.1.1.7 parental virus [49].
Recombination mainly happens in the Receptor Binding Domain (RBD) of the spike protein in the beta coronavirus genus. The statistical analysis supports ancestral recombination. RBD sequence, including two insertions at positions 432-436 and 460-472, is present in both SARS-CoV and SARS-CoV-2, as well as in 427N and 436Y. These variants are helix-shaped and interact with human ACE2 (hACE2) receptors. Recombination involving ancestral strains of SARS-CoV and SARS-CoV-2 and alleles 427N and 436Y increased hACE2 binding affinity, attributing to variations in the sequences [50-57], with the resurgence of novel SARS-CoV-2 variants, which have been detailed elsewhere [58-61].
1.3. Over 10 Variants have Emerged ever Since COVID-19
Although research has shown that recombinational exchanges occur at random along the coronaviral genome, in nature, they are vastly overrepresented in regions controlling viral interaction with host cells [62]. Mutations and recombinations in the genetic material of SARS-CoV-2 can change the virus's life span, infectivity, tropism, and virulence. To introduce a new vaccine to the public domain, it has to go through several steps, from manufacturing to marketing [63]. Although SARS-CoV-2 is rapidly developing, its genome is extremely similar to that of the original Wuhan strain. The dominant delta variant and the discovered recombinant variant in the study share the same spike protein, proving that infectivity and immune evasion are not new properties. Such pronounced genetic changes must be thoroughly investigated, and new variants must be characterized when lineage and clade assignment has been confirmed [64]. The alpha and beta forms of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) isolates from clinical samples from Japan exhibited notable recombination. Such pronounced genetic changes must be thoroughly investigated, and emergent variants must be characterized after meticulously verifying their lineage and clade assignment [65]. Using amplicon-based next-generation sequencing (NGS), a genetically distinct co-infection with two SARS-CoV-2 viruses in a single patient sample was identified in Hungary [66]. Natural selection based on enhanced infectivity and resistance results from viral recombination events within species and across hosts [67, 68]. Despite having relatively similar overall genomic layouts, the 2019-nCoV, human, and bat, SARS-CoV developed into two different groups with different receptor entry specificities as a result of possible recombination in the receptor binding regions. Additionally, 2019-nCoV has a four-amino-acid insertion between the spike protein's S1 and S2 domains that may be cut by furin or TMPRSS2 [69]. The zoonotic potential of coronaviruses is mainly due to recombination. In coronaviruses, the transcription happens at particular sites called transcriptional regulatory sequences (TRS). Recombination between pathogens isolated from different hosts can be explained with a phylogenetic tree [70, 71]. According to our preliminary examination of the SARS-CoV-2 genomes, recombination has the same mechanism as transcription, and recombination is the primary factor for transmission, pathogenicity, and host adaptation. SARS-CoV-2 has a strong recombination ability, which allows it to produce new strains and, in turn, new variants [72]. The last 33 months have seen over 10 variants emerge, with alpha, beta, epsilon, gamma, delta, deltacron, omicron, XE, and XF variants rapidly affecting humans (Fig. 1). After the virus enters the target cell, the membrane fusion domain should be activated. This is accomplished by the target cell protease, which cleaves the S protein into S1 and S2 subunits, thereby activating it. The serine protease TMPRSS2 is used as a protein primer. (In its native state, the SARS-CoV S protein exists as an inactive precursor) [73].
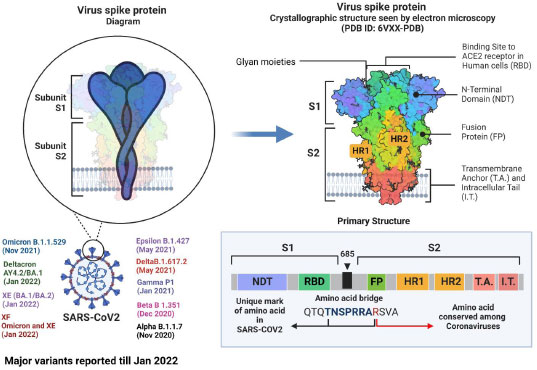
The SARS-CoV-2 genome has 23 mutations, including the critical N501Y mutation in the RBD region of the spike protein [74], The N501Y mutation has mostly been discovered to boost the human ACE2 receptor binding to the SARS-CoV-2 spike protein, which directly contributes to this variation's contagiousness. 9 A P681H mutation near the S1/S2 furin cleavage site of the B.1.1.7 variant could change the rate of S1/S2 dissociation and the binding efficiency to the target cell membrane [74]. Beta variants have E484K and N501Y mutations in common with alpha variants. The variant is crucial because it can evade the host antibodies that recognize both the Receptor Binding Domain and the N-terminal regions of the spike protein [75]. Similarly, three critical mutations exist in the gamma variant (K417T, E484K, and N501Y) found within the S protein Receptor Binding Domain. The ability of an antibody (from natural infection or vaccination) to identify and eradicate the virus is adversely affected by gamma variations. The E484K spike mutation has been linked to patients diagnosed with SARS-CoV-2 re-infection, necessitating additional research into these mutations to develop effective vaccines and antibody-based treatments [76]. The mutations in spike proteins reported are A67V, Δ69-70, T95I, G142D, Δ143-145, Δ211, L212I, ins214EPE, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A, Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F [77]. These factors, in association with antibody evasion, may also have supported its dominance [78].
The delta/B.1.617.2 genome contains 13 mutations (15 or 17, according to other sources, depending on whether more prevalent mutations are included).) that cause changes in the amino-acid sequences of the proteins it encodes. The delta strains are the other variants of concern, formed by additional mutations in the genome that make the protein three-dimensional distinct from the mother variant. The major variations observed in spike proteins of the delta variants are T19R, (G142D), Δ156-157, R158G, L452R, T478K, D614G, P681R, and D950N. The substitution at position 614, the major variation D614G, affects the infectivity and immunogenicity of the virus and is common in many variants of SARS-CoV2 reported so far. In T478K, the Tyr residue at position 478 is replaced by a Lys. Similarly, L452R substitution results in a diminished immune system's capacity for identification and a greater affinity of the spike protein for the ACE2 receptor. It is thought that the P681R mutation will make the variant more infectious at the cell level by allowing the S precursor protein to be cut into the active S1/S2 structure [79]. Recipients with a low antibody response are more susceptible to reinfection, whereas those with a good antibody response have a sterilized immunity. These results demonstrate that molecular signatures linked to vaccine-induced immune responses could be exploited to create biomarkers for the development of vaccine strategies [80]. Viruses can be studied at several levels of biological organization, ranging from the fundamental processes of genome replication, gene expression, and encapsidation to worldwide pandemics. All of these levels are distinct, but they are linked by the presence of threshold conditions that permit the development of a capsid, the loss of genetic information, or the spread of an epidemic [81].
Over the last 12 months, there was a sudden increase in covid cases in China and Asia Pacific with BF.7 could have had these mutations in combination with either all or maybe any three with R346T as the common among all recombination events. However, the Omicron BF.7 lineage has been attributed to infections and is deterministically pathogenic in some countries, such as China not in India [82]. This could be because of the epigenetic spectrum and herd immunity associated with those countries, along with a diffident diet regimen in those countries. On the other hand, the omicron subvariant XBB’s immune evading mutations in BF.7 in combination with R346T, K444T, F486S, and D1199N are known and may have escaped immunity [83] (Table 1).
| S.No. | NCBI Accession | Lineage | Important Mutations | Putative Variant |
|---|---|---|---|---|
| 1 | -MZ780476.1- | B.1.351 | L18F,D80A,D215G,R246I,K417N,E484K,N501Y,D614G,A701V | Beta |
| 2 | -MZ433432.1- | B.1.351 | L18F,D80A,D215G,R246I,K417N,E484K,N501Y,D614G,A701V | Beta |
| 3 | -CAJZLP020000001.1- | P.2 | E484K, D614G,V1176F | Zeta |
| 4 | -CAJFWK010000002.1- | P.2 | E484K, D614G,V1176F | Zeta |
| 5 | -MW562722.1- | B.1.1.7 &Q lineages | N501Y | Alpha |
| 6 | -MT671817.1- | B.1.1.7 | A570D,P681H,T716I,S982A,D1118H | Alpha |
| 7 | -MZ297238.1- | XD | - | Deltacron |
| 8 | -MZ558096.1- | XD | - | Deltacron |
| 9 | -MZ043010.1- | B.1.1.529, BA.1, BA.1.1, BA.2 | A67V, del69-70, T95I, del142-144, Y145D, del211, L212I, ins214EPE, G339D, S371L, S373P, S375F, K417N, N440K, G446S, S477N, T478K, E484A | Omicron |
| 10 | -OM065378.1- | BA.5,BA.3, BA.4 | Q493R, G496S, Q498R, N501Y, Y505H, T547K, D614G, H655Y, N679K, P681H, N764K, D796Y, N856K, Q954H, N969K, L981F | Omicron |
| 11 | -MZ427312.1- | P.1 | E484K, K417T & N501Y | Gamma |
| 12 | -MW621433.1- | P.1 | L18F,T20N,P26S,D138Y,R190S,K417T,E484K,N501Y,H 655Y,T1027I | Gamma |
| 13 | -MZ283644.1- | P.1 | L18F,T20N,P26S,D138Y,R190S,K417T,E484K,N501Y,H 655Y,T1027I | Gamma |
| 14 | -ON017446.1- | P.2 | E484K, D614G,V1176F | Zeta |
| 15 | -ON017450.1- | P.2 | E484K, D614G,V1176F | Zeta |
| 16 | -MZ297238.1- | B.1.617.2 & AY lineages | G142D,T19R,R158G,L452R,T478K,D614G,P681R,D950N,E156del,F157del | Delta |
| 17 | -OK189649.1- | B.1.617.2 and AY lineages | D614G,T478K,L452R,P681R | Delta |
| 18 | -OU577055.1- | B.1.617.2 and AY lineages | D614G,T478K,L452R,P681R | Delta |
CONCLUSION
COVID-19 has been the century’s most destructive pandemic. As the variants of concern and variants of significance are heralding nature, there is an inherent need to bring collaborative convergence among scientists so that we could combat future pandemics such as COVID-19. A question remains about whether these emerging variants could allow us to define the advent of pathogenesis and whether or not we are prepared! This could only be established in the near future.
LIST OF ABBREVIATIONS
| CUB | = Codon Usage Bias |
| NGS | = Next-Generation Sequencing |
| ACE2 | = Angiotensin-Converting Enzyme 2 |
| DDP4 | = Dipeptidyl Peptidase 4 |
| RBD | = Receptor Binding Domain |
| TRS | = Transcriptional Regulatory Sequences |
CONSENT FOR PUBLICATION
Not applicable.
AVAILABILITY OF DATA AND MATERIALS
The data supporting the findings of the article is taken from http://www.ncbi.nlm.nih.gov and gisaid.org.
FUNDING
None.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
ACKNOWLEDGEMENTS
Declared none.

